Rhumatologie
Réunion annuelle de 2019 de l’ACR et de l’ARP
Maîtrise prolongée de la polyarthrite rhumatoïde (PR) : un nouvel inhibiteur des JAK efficace pendant 48 semaines
Atlanta – Selon les nouvelles données d’un essai multicentrique présentées lors de la réunion de 2019 de l’ACR/ARP, un inhibiteur des JAK à petite molécule et à prise orale permet de mieux maîtriser la polyarthrite rhumatoïde (PR) modérée ou grave pendant 48 semaines qu’un inhibiteur du TNF. Ce constat est important puisqu’aucun agent administré par voie orale à des doses types n’avait encore fait preuve d’une maîtrise plus efficace et durable de la PR qu’un agent biologique dans un essai comparatif à répartition aléatoire.
Données à 48 semaines de l’essai SELECT-COMPARE
L’essai SELECT-COMPARE, qui a servi à comparer directement l’upadacitinib, un nouvel inhibiteur des JAK récemment homologué aux États-Unis, à l’adalimumab, un agent biologique, fait partie d’un vaste programme d’essais de phase III sur l’upadacitinib dont les données actualisées ont été présentées lors de la réunion de 2019 de l’ACR/ARP. D’après le chercheur principal, le Dr Roy Fleischmann, de l’École de médecine de l’Université du Sud-ouest du Texas, à Dallas, l’upadacitinib s’est démarqué de l’adalimumab pour beaucoup de paramètres cliniques à 48 semaines alors que les patients des deux groupes recevaient des doses stables de méthotrexate.
Pour une entrevue exclusive avec le Dr Louis Bessette sur l’impact sur la pratique clinique, cliquez ici
Dans l’essai SELECT-COMPARE, 1629 patients dont la maladie risquait énormément d’évoluer ont été répartis aléatoirement dans un rapport de 2:2:1 de façon à recevoir 15 mg d’upadacitinib une fois par jour ou 40 mg d’adalimumab une semaine sur deux ou un placebo pour l’analyse initiale réalisée à 26 semaines. Tous les patients ont continué de suivre leur traitement de fond par le méthotrexate à dose stable.
Les réponses fonctionnelles et cliniques obtenues avec l’inhibiteur de JAK étaient bien supérieures à celles observées avec l’agent biologique lorsque ces deux produits étaient pris avec du méthotrexate.
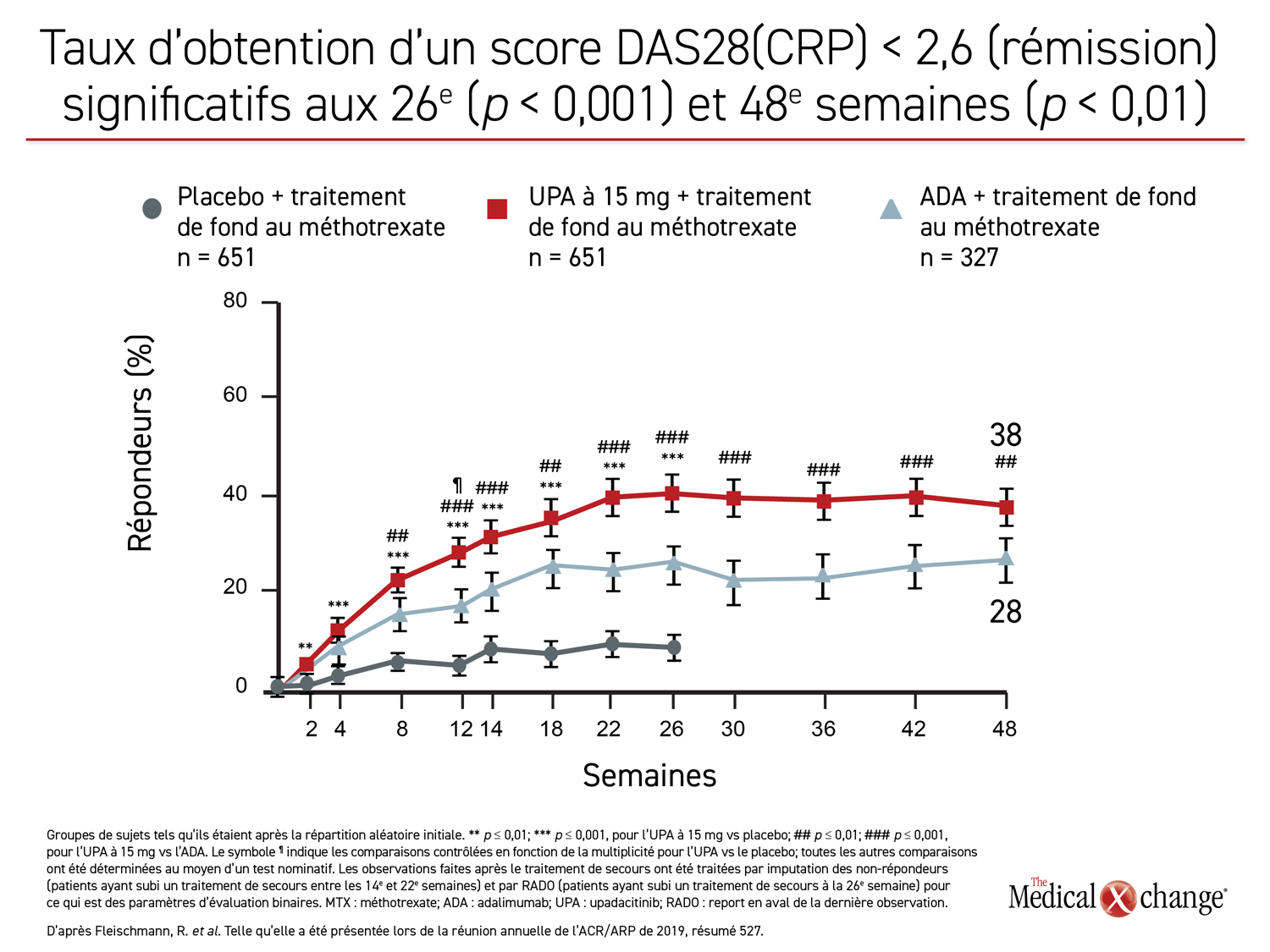
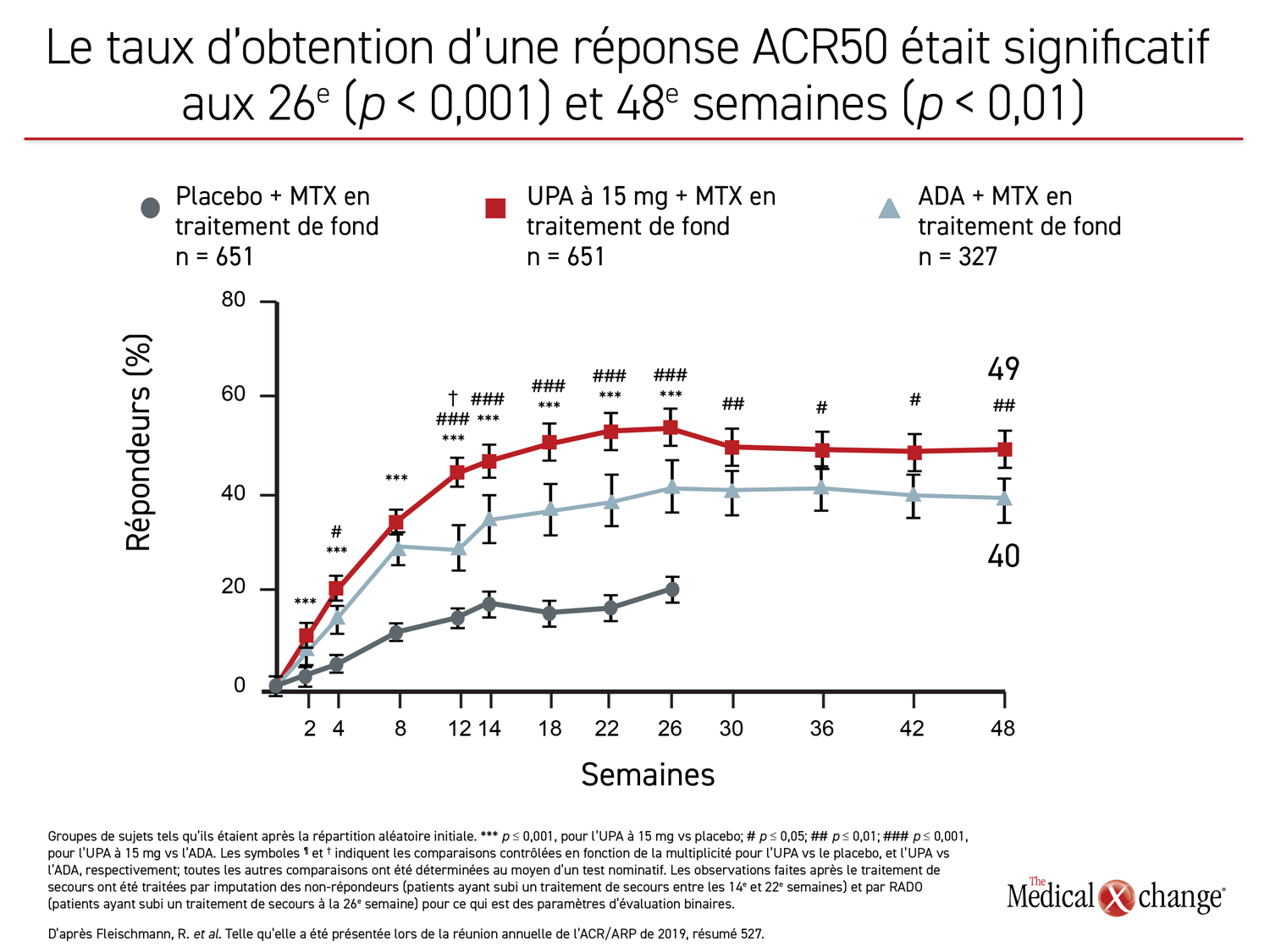
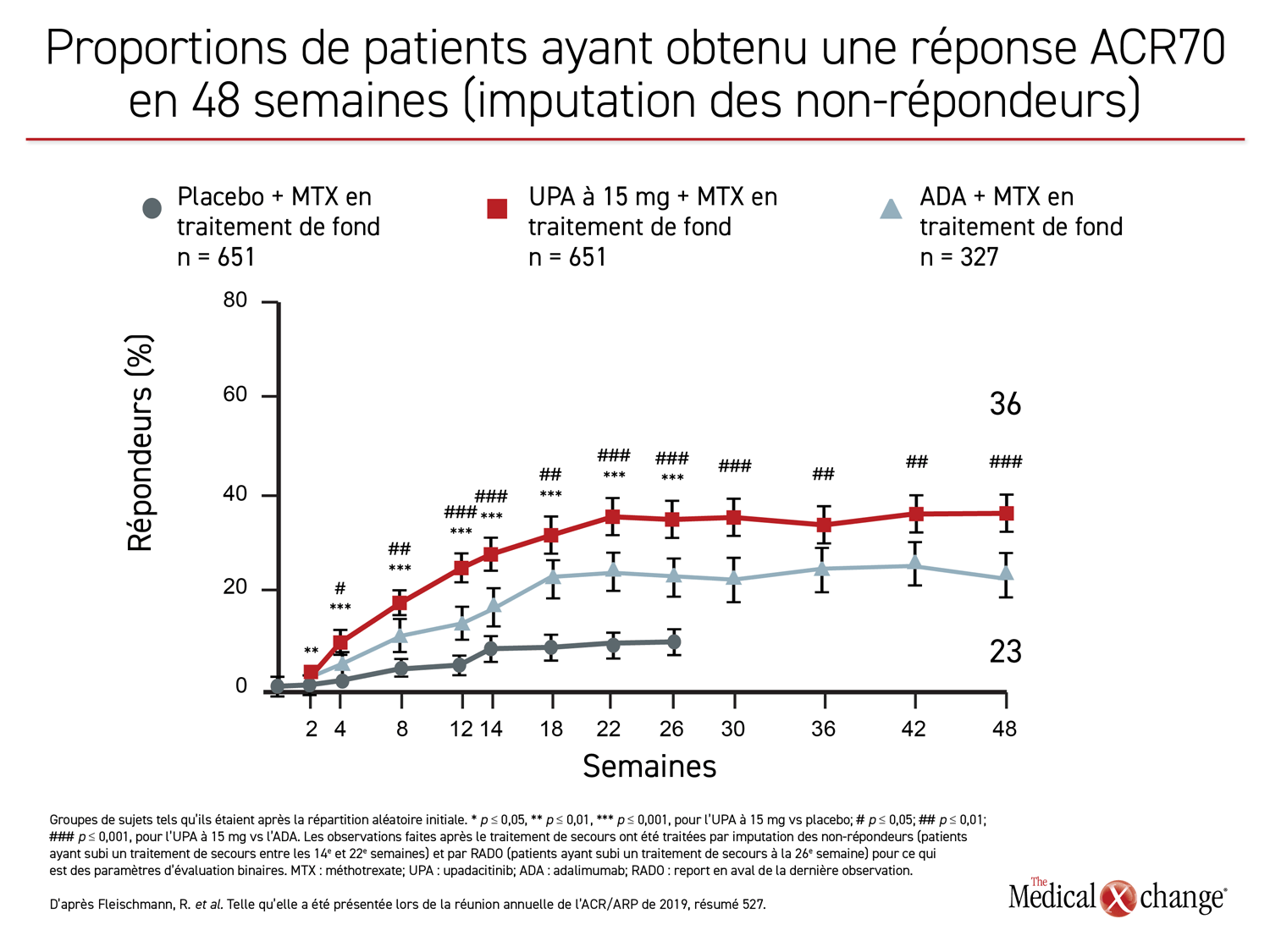
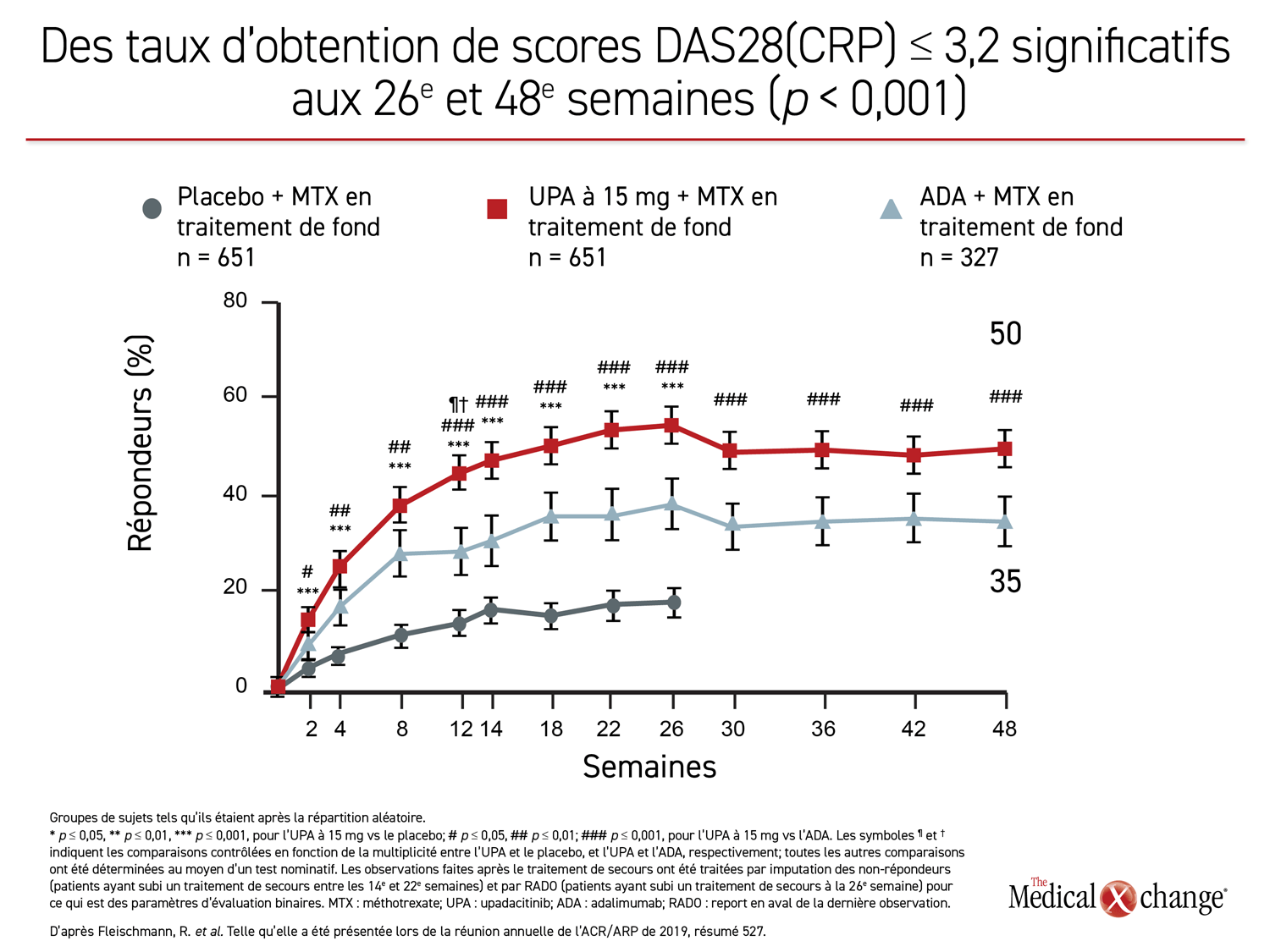
Les données collectées à 26 semaines révélant une meilleure maîtrise de la maladie avec l’upadacitinib ont déjà été publiées (Fleischmann, R. et al. Arthritis Rheumatol 2019;11:1788-1800). L’analyse des données à 48 semaines présentée dernièrement, période pendant laquelle le mode à double insu a été préservé, était prévue au protocole original. Or les résultats obtenus à 48 semaines et ceux rapportés à 26 semaines étaient remarquablement similaires. L’upadacitinib a permis d’obtenir de bien meilleurs résultats pour chacun des critères de mesure utilisés, dont les taux de réponse ACR20, ACR50 et ACR70, ainsi que les taux d’obtention de scores DAS28(CRP) < 2,6 et DAS28(CRP) ≤ 3,2, d’indices CDAI ≤ 10, CDAI ≤ 2,6, SDAI ≤ 3,3 et l’indice HAQ-DI.
Bien des écarts entre les groupes de traitement actif étaient déjà marqués à 12 semaines. Or les données collectées à 48 semaines ne montrent aucun fléchissement de l’avantage relatif de l’upadacitinib sur l’adalimumab jusqu’à la rencontre de suivi la plus récente. Tous les écarts entre ces agents qui étaient statistiquement significatifs à 26 semaines, notamment pour le taux de réponse ACR70 et ACR50, et le taux d’obtention de scores DAS28(CRP) < 2,6 l’étaient toujours à 48 semaines (figure 1), (figure 2).
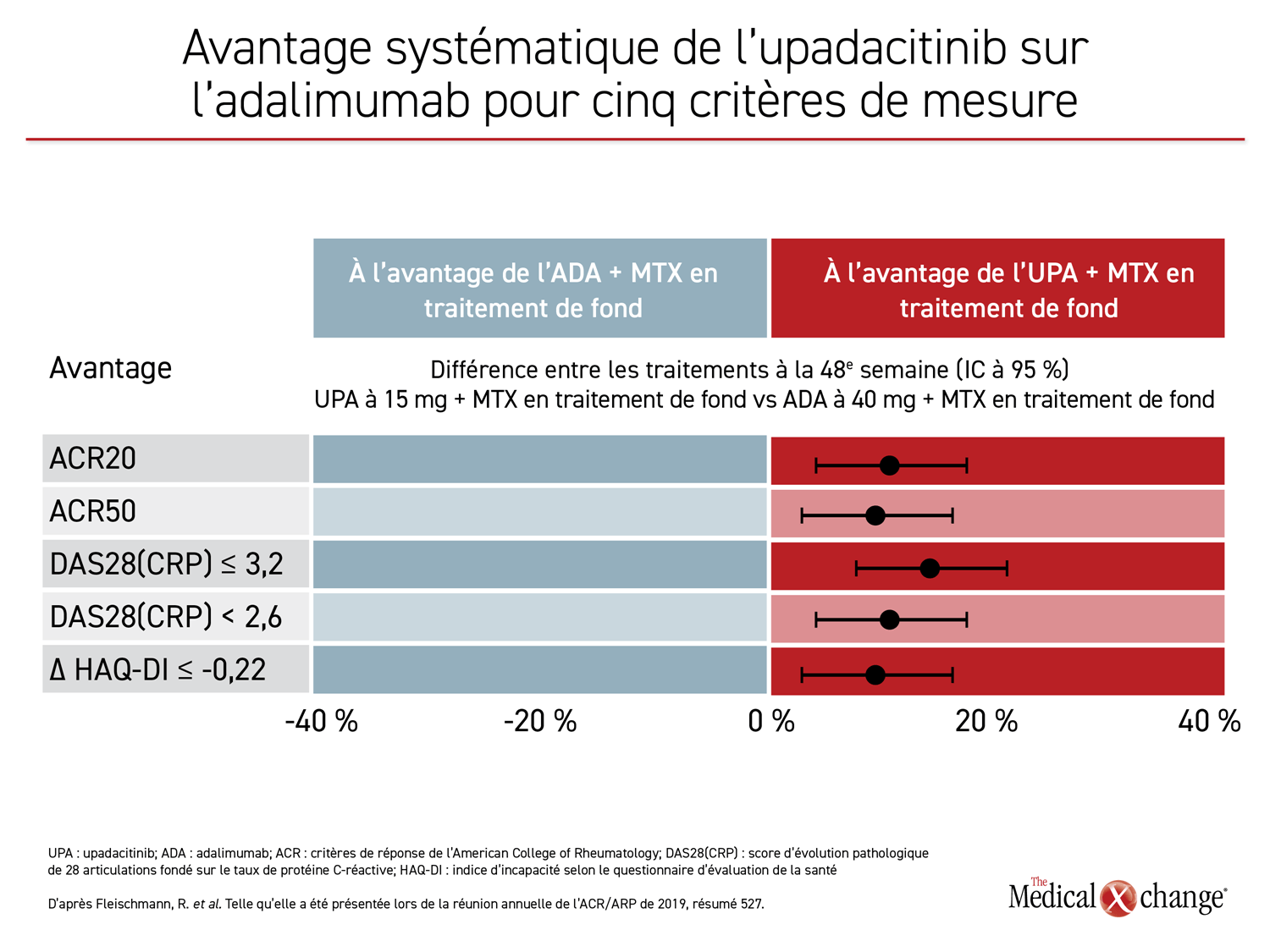
L’avantage relatif de l’upadacitinib sur l’adalimumab était remarquablement similaire pour chacun des paramètres d’évaluation importants, ce qui confirme son effet anti-inflammatoire plus profond et plus durable (figure 3).
Même s’il y avait des différences entre le bilan d’innocuité de l’upadacitinib et celui de l’adalimumab, il n’empêche que la proportion de patients victimes d’un effet indésirable sérieux (12,9 % vs 15,6 %) ou ayant motivé l’abandon du traitement (7,4 % vs 11,1 %) était plus faible à la 48e semaine dans le groupe affecté à l’upadacitinib. La proportion de patients ayant présenté un zona de novo était plus forte chez ceux traités par l’inhibiteur de JAK que chez ceux qui avaient reçu l’agent biologique (3,1 % vs 1,3 %), mais le taux de thromboembolie veineuse (TEV) était plus faible (0,3 vs 1,1).
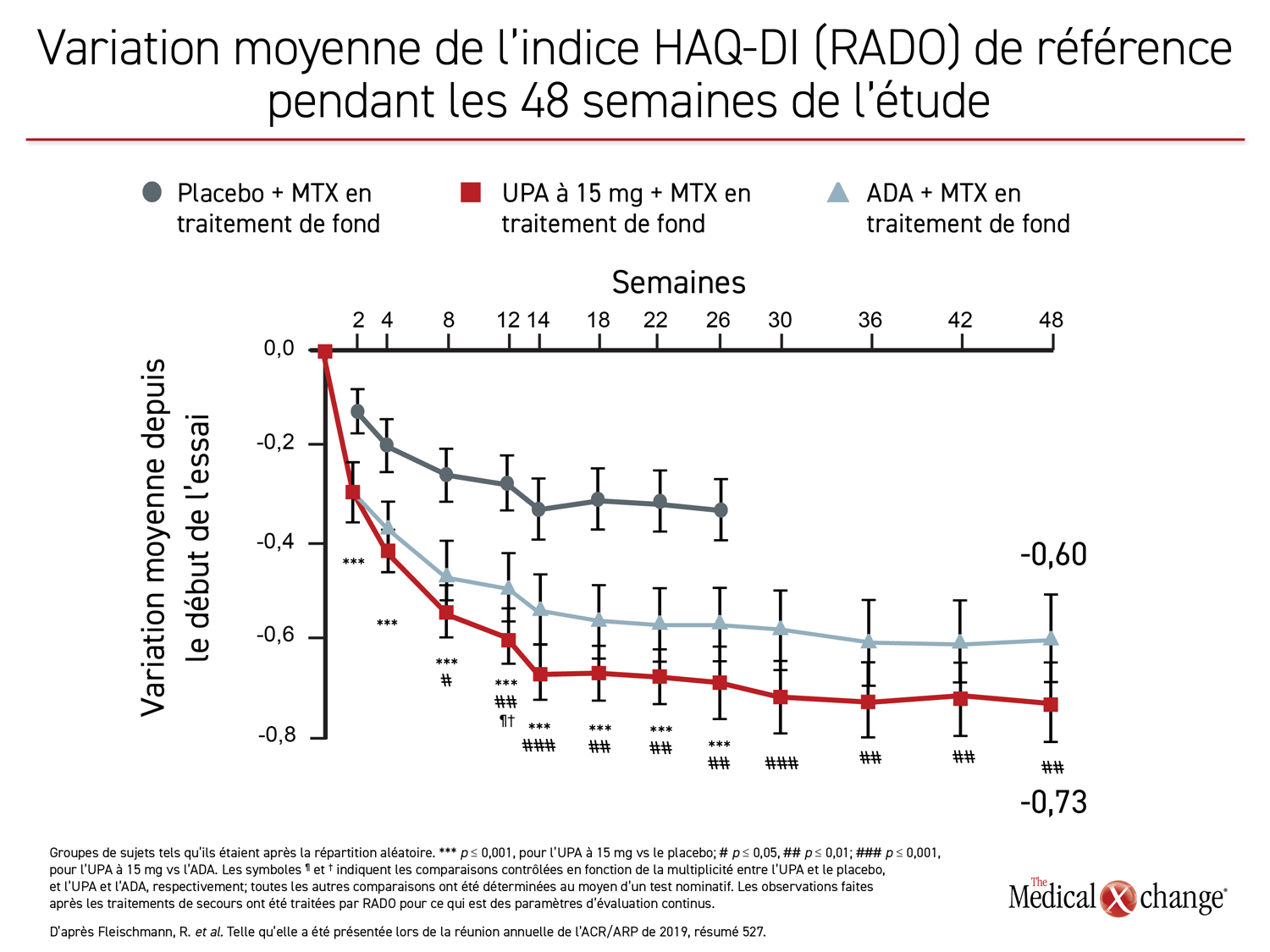
En plus des critères de mesure standard de l’activité inflammatoire, tels qu’un score DAS28(CRP) < 2,6 et un indice CDAI ≤ 10, les patients traités par l’upadacitinib ont vécu une plus belle expérience que ceux traités par l’adalimumab comme en témoignent l’indice HAQ-DI et les scores attribués à la douleur. Pour ce qui est de l’indice HAQ-DI, l’upadacitinib a nettement eu l’avantage sur l’adalimumab dès la 8e semaine. Sur le plan statistique, l’avantage observé à la 48e semaine équivalait à celui objectivé à la 26e semaine (figure 4). Quant à la douleur rapportée par les patients, l’avantage de l’upadacitinib avait atteint le seuil de la signification statistique à la 4e semaine. Encore là, l’avantage relatif de l’upadacitinib était de la même ampleur aux 26e et 48e semaines.
L’évolution de l’atteinte structurelle dans les deux groupes de traitement actif n’a pas été comparée au moyen de radiographies, mais le groupe upadacitinib a été comparé au groupe placebo. Selon le Dr Fleischmann : « L’inhibition importante de l’atteinte articulaire structurelle observée à la 26e semaine avec l’upadacitinib à 15 mg par rapport à celle obtenue avec le placebo était toujours présente à la 48e semaine ».
Les patients ayant reçu de l’upadacitinib dès le départ étaient moins susceptibles d’avoir eu besoin d’un traitement de secours avant la fin des 48 semaines que ceux traités par l’adalimumab (38,7 % vs 48,6 %). Chez les patients qui ont poursuivi le traitement auquel ils avaient été affectés au-delà de la 26e semaine, 86 % de ceux traités par l’upadacitinib prenaient toujours cet agent à la 48e semaine par rapport à 76 % de ceux prenant de l’adalimumab.
Autres comparaisons entre un inhibiteur de JAK et un agent biologique
Les comparaisons directes peuvent être intéressantes lorsqu’il faut trouver une solution aux traitements de fond de synthèse classiques ne permettant pas de bien maîtriser la PR. Bien que l’essai SELECT-COMPARE ait été le premier essai multicentrique ayant fait ressortir l’avantage significatif d’une dose type d’un inhibiteur de JAK sur un agent biologique, il n’est pas le premier qui ait reposé sur une comparaison directe. En effet, le tofacitinib, le premier inhibiteur de JAK qui ait été homologué, et le baricitinib ont été comparés à l’adalimumab au cours d’essais contrôlés par placebo ayant des plans similaires.
Lors de l’essai sur le tofacitinib, 717 patients ont été répartis aléatoirement de façon à recevoir une des deux doses de cet agent (5 ou 10 mg, 2 fois par jour), de l’adalimumab ou un placebo (van Vollenhoven, R. et al. N Engl J Med 2012;367:508-519). Comme ce fut le cas pendant l’essai SELECT-COMPARE, tous les patients ont poursuivi leur traitement de fond par le méthotrexate à dose stable. À six mois, les trois groupes de traitement actif ont obtenu de meilleurs résultats que le groupe placebo pour les trois principaux critères de mesure, soit le taux de réponse ACR20 et de scores DAS28(CRP) < 2,6, et l’indice HAQ-DI, mais il n’y avait pas de différence significative entre eux.
Pendant l’essai sur le baricitinib, 1307 patients ont été affectés aléatoirement à du baricitinib à 4 mg une fois par jour, à de l’adalimumab ou à un placebo (Taylor, P.C. et al. N Engl J Med 2017;376:652-662). La dose de 4 mg de baricitinib a eu un avantage statistique par rapport à l’adalimumab pour le paramètre d’évaluation principal, soit le taux de réponse ACR20 à la 12e semaine (70 % vs 61 %; p = 0,014), mais elle est plus élevée que celle qui est homologuée au Canada pour le traitement de la PR. De plus, les écarts enregistrés pour ce critère n’étaient pas significatifs aux 16e et 32e semaines.
Différences entre les inhibiteurs de JAK
Plusieurs raisons expliquent que les inhibiteurs de JAK agissent différemment, mais la détermination de l’ampleur de la sélectivité de chacun pour les JAK présente un intérêt particulier, puisque des quatre voies de transduction des JAK (JAK1, JAK2, JAK3 et Tyk2), c’est JAK1 qui a été la plus fortement incriminée dans l’activité pro-inflammatoire. Alors que le tofacitinib inhibe ces quatre voies, l’upadacitinib et le baricitinib font preuve d’une plus grande sélectivité pour JAK1 que pour JAK3 en contexte expérimental. Contrairement à l’upadacitinib, le baricitinib affiche une activité similaire pour JAK2. La possibilité que ces différences de sélectivité influencent le rapport avantages-risques du traitement de la PR est en cours d’évaluation.
« Rien n’indique que des doses plus fortes permettent d’obtenir de meilleurs résultats, du moins dans le traitement de la PR », a expliqué le Dr Gerd R. Burmester, de l’Hôpital universitaire de la Charité, à Berlin, en Allemagne, et auteur principal de la mise à jour de l’essai SELECT-NEXT, qui servait au départ à comparer l’upadacitinib à 15 mg, l’upadacitinib à 30 mg et un placebo pendant 12 semaines chez des patients ne répondant pas suffisamment aux traitements de fond de synthèse classiques. Pendant la réunion, il a parlé des données recueillies pendant la phase de prolongation de 60 semaines menée à l’insu.
Efficacité similaire des doses de 15 mg et de 30 mg
Sur les 661 patients recrutés au départ dans l’essai SELECT-NEXT, 618 ont participé à la phase de prolongation. À ce moment-là, les patients recevant le placebo sont passés à l’un des groupes traités par l’upadacitinib. Or à la 60e semaine, les taux de réponse restaient élevés au vu de nombreux critères de mesure. Ces réponses étaient comparables que les sujets soient passés du placebo à un des traitements actifs ou qu’ils aient été affectés d’emblée à la dose de 15 ou de 30 mg d’upadacitinib. Par exemple, environ 70 % des sujets ayant pris l’une ou l’autre de ces doses ont obtenu une réponse ACR50, peu importe qu’ils l’aient prise dès le départ ou qu’ils y soient passés par la suite.
En outre, 80 % des participants à la phase de prolongation suivaient toujours leur traitement à la 60e semaine et seulement 10 (1,6 %) de ceux ayant abandonné en cours de route l’ont fait pour cause d’un manque d’efficacité et 50 (8,2 %), en raison d’un effet indésirable.
« Comme la dose plus forte n’offre pas plus d’efficacité, le choix de la dose de 15 mg est plus logique. »
« Le taux d’abandon a été un peu plus élevé dans le groupe à 30 mg, mais nous n’avons pas noté de différence pour ce qui est des effets indésirables motivant ces abandons », a affirmé le Dr Burmester. Même si la dose de 30 mg a été bien tolérée, il a ajouté : « Comme la dose plus forte n’offre pas plus d’efficacité, le choix de la dose de 15 mg est plus logique ».
Ce constat est différent de celui établi avec le tofacitinib et le baricitinib qui se sont révélés plus efficaces aux doses élevées, quoiqu’assortis de plus d’effets indésirables.
Évaluation du risque de TEV et d’incidents cardiovasculaires majeurs (ICVM)
Reste à savoir si ces agents en particulier et les inhibiteurs de JAK en général augmentent le risque de TEV, qui est déjà élevé chez les patients aux prises avec la PR, et le cas échéant, dans quelle mesure. Il ressort de l’analyse d’une base de données où sont répertoriés plus de 30 000 patients atteints de cette maladie, que le risque de TEV est élevé numériquement parlant, mais que la différence enregistrée entre ceux traités par le tofacitinib et ceux ayant reçu un anti-TNF n’est pas statistiquement significative (Desai, R.J. et al. Arthritis Rheumatol 2019;781:892-900). Les données groupées de neuf études sur le baricitinib opposé à la PR n’ont révélé aucune différence entre l’incidence à long terme des TEV et des incidents cardiovasculaires majeurs (ICVM) et les taux rapportés antérieurement chez les patients atteints de cette maladie (Taylor, P.C. et al. Arthritis Rheumatol 2019;71:1042-1055).
Les taux de TEV et d’ICVM étaient comparables et correspondaient à ceux généralement rapportés chez les patients atteints de PR.
L’analyse des données du programme d’essais cliniques de phase III SELECT consacré à l’upadacitinib, qui a été présentée lors de la réunion de 2019 de l’ACR/ARP, ne laisse entrevoir aucune hausse du risque de TEV ou d’ICVM par rapport à celui observé avec le placebo, même lorsqu’elle se limite aux patients ayant reçu la dose de 30 mg. Cette analyse n’a pas porté uniquement sur les essais SELECT-NEXT et SELECT-BEYOND, qui comporte des données s’étendant sur 60 semaines, mais aussi sur trois autres études.
« La fréquence des incidents, qui était similaire dans les groupes de traitement actif et les groupes placebos, se comparait à celle généralement rapportée chez les patients atteints de PR », a déclaré le Dr Ernest Choy, de l’École de médecine de l’université de Cardiff, au R.-U. La comparaison des taux de TEV et d’ICVM enregistrés avec l’upadacitinib à 15 mg, l’upadacitinib à 30 mg, le placebo et l’adalimumab a permis de constater que les intervalles de confiance se chevauchaient parfaitement.
On ignore quelles peuvent être les différences, le cas échéant, entre les agents appartenant à la classe des inhibiteurs à petite molécule et à celle des agents biologiques ou à l’intérieur de ces classes. Quoique rassurante, l’absence du moindre signe préoccupant, même à la dose la plus élevée d’upadacitinib, est actuellement étudiée de façon plus approfondie.
Conclusion
Aujourd’hui, les inhibiteurs des JAK sont très souvent utilisés avant les agents biologiques chez les patients atteints de PR réfractaire aux traitements de fond de synthèse classiques, surtout en raison de la préférence pour les agents à prise orale. Si les études antérieures donnent à penser qu’ils sont aussi efficaces que les agents biologiques, il n’empêche que l’upadacitinib est le premier agent de cette classe à s’être montré plus efficace pour plusieurs paramètres d’évaluation sur une période de 48 semaines. S’il est homologué au Canada, l’upadacitinib sera sans doute un ajout majeur à l’arsenal thérapeutique opposé à la PR avancée.
Diapositives Additionelles