Hématologie
Réunion et exposition annuelles de l’American Society of Hematology (ASH)
Un essai de phase III : nouvelle option thérapeutique prometteuse pour les patients atteints de LLC jamais traitée auparavant
Orlando – Un inhibiteur de la tyrosine kinase de Bruton (BTK) de deuxième génération vient bousculer les options thérapeutiques opposées à la leucémie lymphoïde chronique (LLC). Les données de l’essai ELEVATE-TN présentées à la réunion annuelle de 2019 de l’ASH établissent un parallèle entre l’acalabrutinib, un nouvel inhibiteur sélectif de la BTK utilisé en première intention, et une protection sans précédent contre l’évolution de la maladie ou le décès comparativement à un schéma chimiothérapique. Le gain observé chez les patients jamais traités auparavant vient s’ajouter à l’activité exceptionnelle déjà documentée dans les cas de LLC récidivante ou réfractaire. La FDA a homologué ces deux indications deux semaines à peine avant la réunion.
Il est à prévoir que par rapport à l’ibrutinib, un inhibiteur de la BTK de première génération, l’acalabrutinib tire son avantage clinique de sa plus grande sélectivité pour la BTK ciblée, d’où un effet thérapeutique plus marqué et une activation moins forte des autres kinases. Lors de l’essai de phase III intitulé ELEVATE-TN, l’acalabrutinib allié à de l’obinutuzumab (un anticorps monoclonal anti-CD20) a été utilisé en première intention et comparé au traitement type associant l’obinutuzumab au chlorambucil. Un troisième groupe de sujets a reçu de l’acalabrutinib en monothérapie.
Pour une entrevue exclusive avec le Dr Pierre Laneuville sur l’impact sur la pratique clinique, cliquez ici
Atteinte de l’objectif principal de l’étude : survie sans progression de la maladie
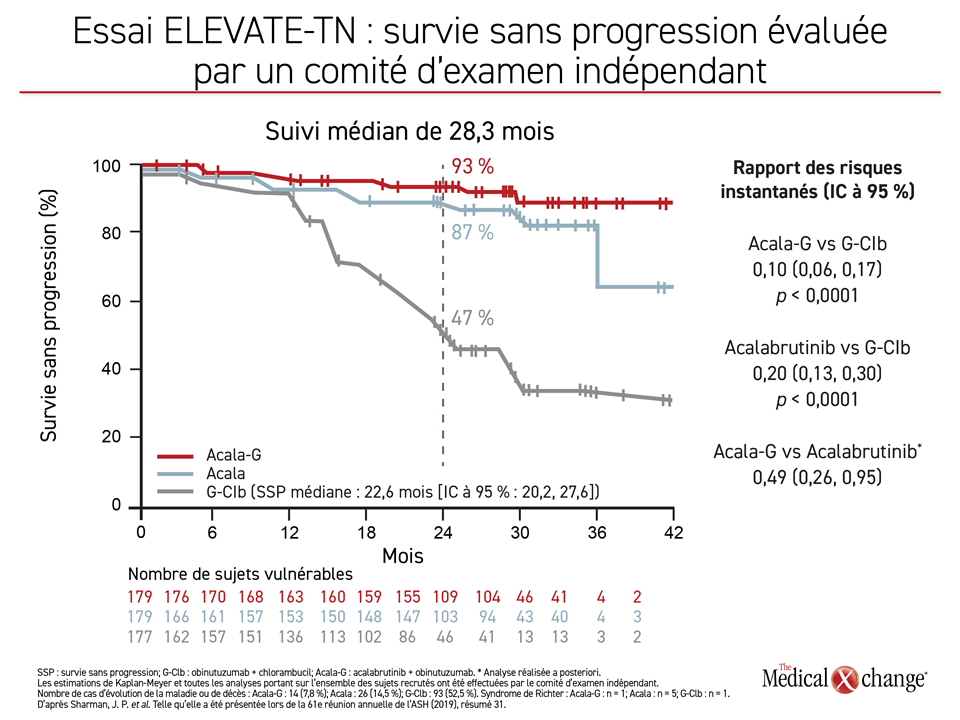
L’objectif principal de cet essai intéressant la survie sans progression (SSP) a été atteint, les réductions dans les taux d’évolution de la maladie ou de décès s’étant révélées statistiquement significatives dans les deux groupes traités par l’acalabrutinib comparativement à la chimiothérapie type (Figure 1).
« Le gain relatif obtenu pour la SSP avec l’acalabrutinib allié ou non à de l’obinutuzumab a systématiquement été observé dans tous les sous-groupes, qu’il y ait présence ou non de caractéristiques très vulnérantes », a rapporté le Dr Jeff P. Sharman, Directeur de la Recherche, du Willamette Valley Cancer Institute, à Eugene, en Orégon.
« Le gain relatif obtenu pour la SSP avec l’acalabrutinib allié ou non à de l’obinutuzumab a systématiquement été observé dans tous les sous-groupes, qu’il y ait présence ou non de caractéristiques très vulnérantes. »
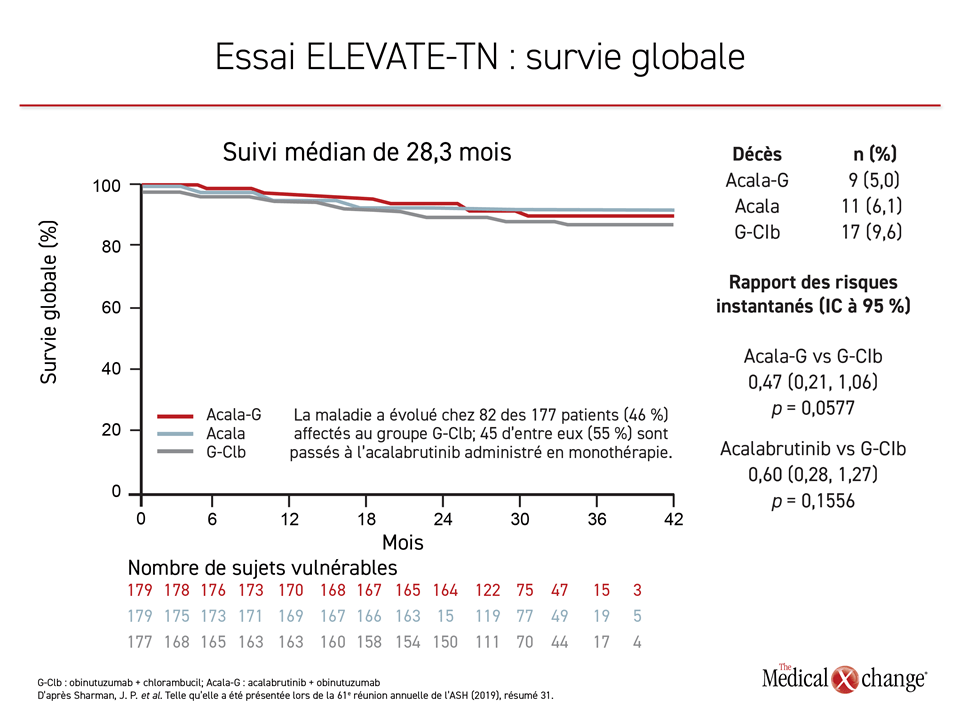
En dépit du fait que le plan de l’essai permettait aux témoins de passer à l’acalabrutinib, les chercheurs ont noté une tendance prometteuse du côté de la survie globale (SG), et ce même au bout d’un suivi médian de 28,3 mois seulement (Figure 2). Selon les estimations, la survie à 30 mois dans le groupe acalabrutinib en association, le groupe acalabrutinib en monothérapie et le groupe chlorambucil-obinutuzumab s’élevait à 95 %, à 94 % et à 90 %.
Le groupe traité en monothérapie y compris
Au cours de l’essai ELEVATE-TN, 535 patients dont la LLC n’avait jamais été traitée ont été répartis aléatoirement en trois groupes : acalabrutinib-obinutuzumab, acalabrutinib en monothérapie et chlorambucil-obinutuzumab. Ce dernier est un traitement type administré en première intention contre la LLC qui a été utilisé dans plusieurs autres essais réalisés récemment en pareil contexte. Selon le Dr Sharman, au terme d’un suivi médian de 28,3 mois, la SSP médiane n’était toujours pas atteinte dans le groupe acalabrutinib-obinutuzumab, alors qu’elle se situait à 22,6 mois dans le groupe obinutuzumab-chlorambucil, d’où une réduction de 90 % (RRI : 0,10; p < 0,0001) des cas d’évolution de la maladie ou de décès par rapport au groupe chlorambucil-obinutuzumab. Dans le groupe traité en monothérapie, une amélioration de la SSP de 80 % a été observée comparativement au groupe traité par chimiothérapie (RRI : 0,20; p < 0,0001). Il reste à déterminer si ces avantages relatifs par rapport au groupe témoin sont différents.
« L’essai n’était pas conçu et n’était pas doté d’une puissance suffisante pour comparer la SSP des deux groupes traités par l’acalabrutinib », a toutefois précisé le Dr Sharman. Résultat : l’éventuelle différence clinique entre les deux est inconnue, mais il demeure que l’essai a permis de constater l’activité importante exercée par l’acalabrutinib qu’il soit administré avec ou sans obinutuzumab. Le rôle de l’acalabrutinib administré en monothérapie a son importance pour ce qui est de la commodité du traitement pour les patients, mais aussi pour les coûts. Il fera sans doute l’objet de nouvelles études.
« L’acalabrutinib allié à l’obinutuzumab a été relié à une baisse de 90 % du risque d’évolution de la maladie ou de décès. »
L’essai ELEVATE-TN met à l’épreuve un traitement sans agent chimiothérapique
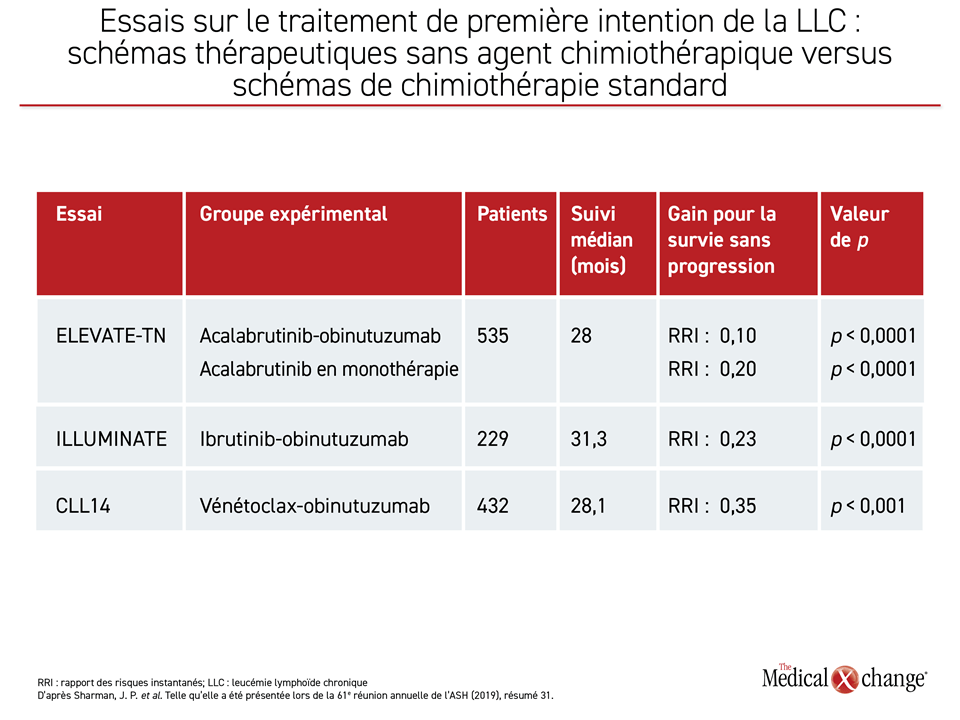
L’essai ELEVATE-TN est le troisième essai majeur de phase III ayant servi à évaluer un schéma thérapeutique sans agent chimiothérapique dans le traitement de première intention de la LLC; il succède aux essais ILLUMINATE et CLL14 menés selon un plan similaire. Lors de ces trois essais, un agent expérimental allié à l’obinutuzumab a été comparé à une association formée de chlorambucil et d’obinutuzumab. L’essai ILLUMINATE a donné lieu au début de l’année à l’homologation de l’ibrutinib par la FDA à titre de traitement de première intention de la LLC. L’essai CLL14 a quant à lui valu au vénétoclax, un inhibiteur de la BCL-2, d’être homologué par la FDA plusieurs mois plus tard.
Tous ces essais ont établi, sous l’angle de la SSP, un parallèle entre le traitement expérimental et un avantage statistiquement significatif par rapport au traitement chimiothérapique standard, fournissant ainsi un important faisceau d’arguments à l’appui des traitements de première intention sans agent chimiothérapique dans les cas de LLC (Tableau 1). Sans comparaison directe, il est impossible de mesurer l’importance de l’avantage relatif plus marqué objectivé pendant l’essai ELEVATE-TN dans l’un ou l’autre des groupes traités par l’acalabrutinib par rapport à la chimiothérapie en regard des résultats obtenus dans les deux autres essais récents.
La tolérabilité de l’acalabrutinib est acceptable
L’acalabrutinib a été bien toléré lors de l’essai ELEVATE-TN, qu’il ait été administré seul ou en association avec l’obinutuzumab. Par ailleurs, le taux d’abandons motivés par un effet indésirable a été plus élevé dans le groupe chlorambucil-obinutuzumab (14,1 %) que dans ceux affectés à l’association acalabrutinib-obinutuzumab (11,2 %) ou à l’acalabrutinib en monothérapie (8,9 %).
Les taux d’abandons pour cause de neutropénie de grade 3 ou plus, qui était l’effet indésirable le plus répandu dans les trois groupes de sujets, s’élevaient respectivement à 29,9 %, à 9,5 % et à 41,4 % dans le groupe acalabrutinib-obinutuzumab, le groupe acalabrutinib en monothérapie et le groupe chlorambucil-obinutuzumab. Les cas de céphalée, de diarrhée et d’arthralgie étaient plus fréquents dans les groupes traités par l’acalabrutinib, mais ils étaient majoritairement bénins. L’incidence des céphalées (1,1 %, 1,1 % et 0 %), de diarrhée (4,5 %, 0,5 % et 1,8 %) et d’arthralgie (1,1 %, 0,6 % et 1,2 %) de grade 3 ou plus était semblable dans les trois groupes de sujets.
Les effets indésirables d’intérêt rapportés
Parmi les effets indésirables d’intérêt, la pneumonie a été plus fréquente dans le groupe traité par l’acalabrutinib en association (6,7 %) ou en monothérapie (2,8 %) que dans le groupe ayant reçu le traitement comprenant un agent chimiothérapique (1,8 %), contrairement à la pneumonie fébrile, qui elle a été plus fréquente dans ce dernier groupe (4,1 % vs < 2 %). Les taux d’infection de grade 3 ou plus (20,8 % et 14,0 % vs 8,2 %) et de saignements (42,7 % et 39,1 % vs 11,8 %) étaient plus élevés dans le groupe traité par l’acalabrutinib en association ou en monothérapie que dans le groupe ayant reçu le traitement comprenant un agent chimiothérapique, mais les saignements majeurs de grade 3 ou plus n’étaient que légèrement plus fréquents (1,7 % et 1,7 % vs 0 %).
De l’avis Dr Sharman, il faut toutefois examiner le risque relatif lié à ces effets indésirables en regard de l’exposition relative. La durée médiane du traitement a été de 27,7 mois chez les patients ayant reçu l’association acalabrutinib-obinutuzumab, celui-ci ayant pu durer jusqu’à 40,2 mois chez certains d’entre eux, alors qu’elle n’a été que de 5,6 mois dans le groupe traité par le schéma comprenant un agent chimiothérapique. La plupart des patients des deux groupes traités par l’acalabrutinib poursuivaient toujours leur traitement au moment où l’insu a été levé.
Selon l’analyse du comité d’examen indépendant, 93,9 % (taux de réponse objective; TRO) des sujets traités par l’acalabrutinib en association ont répondu au traitement, 13 % d’entre eux ayant affiché une réponse complète (RC). Dans le groupe ayant reçu l’acalabrutinib en monothérapie, le TRO a atteint 85,5 %. Dans le groupe chlorambucil-obinutuzumab, 78,5 % (TRO) des sujets ont répondu au traitement, soit un pourcentage significativement inférieur à celui obtenu dans le groupe traité par l’acalabrutinib en association (p < 0,001). Par ailleurs, la proportion de sujets ayant obtenu une RC y était également plus faible.
Un gain constant d’un sous-groupe à l’autre
Qu’il ait été administré seul ou en association avec l’obinutuzumab, l’acalabrutinib a constamment eu l’avantage sur l’association chlorambucil-obinutuzumab pour l’ensemble des sous-groupes prévus au protocole, à preuve le graphique en forêt présenté par le Dr Sharman. Ces sous-groupes étaient formés, entre autres, en fonction du volume tumoral de départ, de caractéristiques cytogénétiques très vulnérantes et du caryotype. L’âge médian des sujets de cet essai était de 70 ans, mais le gain observé chez les sujets de moins de 65 ans était voisin de celui obtenu chez leurs aînés. Le gain relatif obtenu avec l’acalabrutinib était uniforme, que les sujets soient désavantagés sur le plan génétique ou non. Pour la SSP des patients porteurs de la del(17p) par exemple, l’emploi de l’association acalabrutinib-obinutuzumab a eu un avantage estimé à 87 % (RRI : 0,13) sur le schéma comprenant un agent chimiothérapique.
« Contrairement aux observations faites dans certaines études récentes, l’acalabrutinib a permis d’améliorer la SSP globale par rapport aux traitements auxquels il était comparé tant dans les sous-groupes de sujets porteurs d’un gène IgVH muté que non muté [gène codant pour les chaînes lourdes de l’immunoglobuline] », a affirmé le Dr Sharman.
« Contrairement aux observations faites dans certaines études récentes, l’acalabrutinib a permis d’améliorer la SSP globale par rapport aux traitements auxquels il était comparé tant dans les sous-groupes de sujets porteurs d’un gène IgVH muté que non muté. »
Les résultats obtenus avec l’acalabrutinib comme traitement de première intention de la LLC suivent de quelques mois à peine ceux tirés d’un essai de phase III mené chez des patients atteints de LLC récidivante ou réfractaire. Cet essai, intitulé ASCEND, qui a été présenté lors du congrès de 2019 de l’European Hematology Association, a servi à comparer des doses de 100 mg d’acalabrutinib à prise orale administré seul, soit une dose identique à celle utilisée dans les deux groupes expérimentaux de l’essai ELEVATE-TN, au rituximab administré en association avec de l’idélalisib (un inhibiteur sélectif de l’isoforme de la phosphatidylinositol 3-kinase) ou de la bendamustine, le choix étant laissé au chercheur.
L’âge médian des 310 sujets qui y ont été recrutés était de 67 ans et 40 % environ d’entre eux affichaient des caractéristiques cytogénétiques défavorables. Le nombre médian de traitements antérieurs se chiffrait à 1, mais certains des patients recrutés avaient déjà subi jusqu’à 8 traitements.
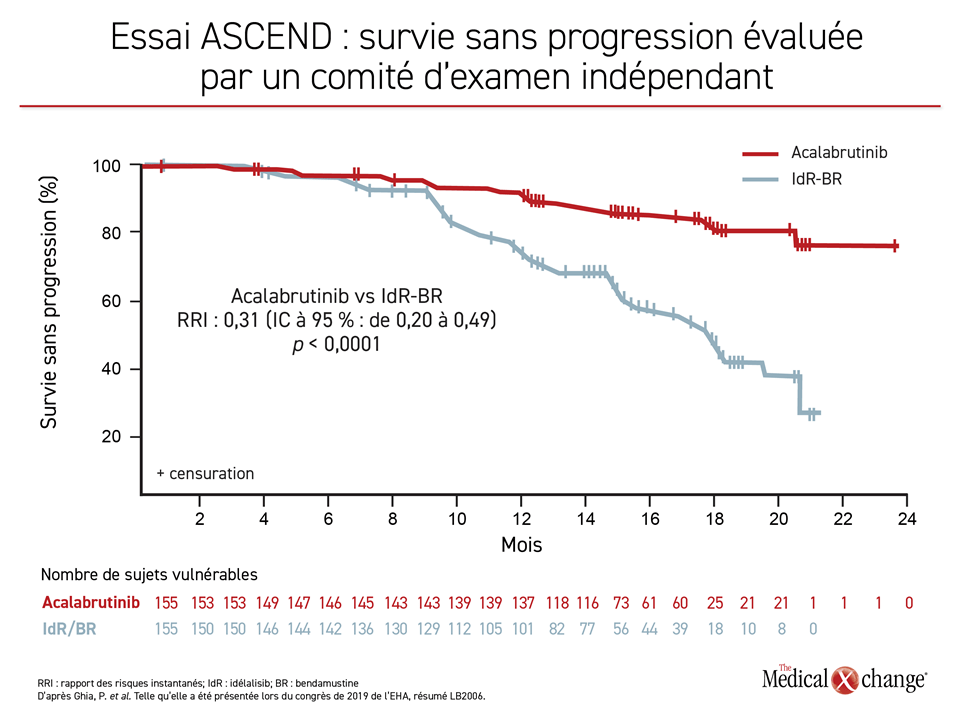
Taux de SSP dans les cas de LLC récidivante ou réfractaire : 88 % vs 68 %
Les taux de SSP à 12 mois enregistrés pendant l’essai ASCEND s’élevaient à 88 % dans le groupe affecté aléatoirement à l’acalabrutinib à prise orale administré seul comparativement à 68 % avec l’une ou l’autre des associations médicamenteuses, d’où une protection relative de 69 % contre l’évolution de la maladie et le décès (RRI : 0,31; p < 0,0001) (Figure 3). Encore là, en dépit du fait que le protocole permettait de passer à l’acalabrutinib, ce qu’on fait 23 % des patients qui au départ avaient été affectés aux associations médicamenteuses, les taux de SSP (88 % vs 68 %) et de SG (94 % vs 91 %) à 12 mois étaient quand même plus élevés dans le groupe acalabrutinib.
Dans les cas de LLC récidivante ou réfractaire, l’acalabrutinib s’est montré supérieur aux associations médicamenteuses dans tous les sous-groupes, y compris ceux formés en fonction de caractéristiques cytogénétiques défavorables.
D’après le Dr Paolo Ghia, chercheur principal et Directeur du programme de recherche stratégique sur la LLC, à l’Université Vita-Salute San Raffaele, de Milan, en Italie, l’amélioration relative de la SSP obtenue avec l’acalabrutinib lors de l’essai ASCEND s’est produite dans tous les sous-groupes, y compris ceux formés en fonction de caractéristiques cytogénétiques défavorables ou de facteurs de risque, tels que les mutations de TP53 et le stade III de Rai ou plus, comme ce fut le cas d’ailleurs pendant l’essai ELEVATE-TN.
De plus, les sujets ont mieux supporté l’acalabrutinib. À l’instar de ce qui a été constaté pendant l’essai ELEVATE-TN, les effets indésirables les plus souvent observés chez les participants à l’essai ASCEND traités par l’inhibiteur de la BTK de deuxième génération, qui étaient bénins pour la plupart, ont été les céphalées (22 %), la neutropénie (19 %) et la diarrhée (18 %). Les effets indésirables de grade 3 ou plus observés avec la plus grande fréquence chez les sujets traités par l’acalabrutinib étaient la neutropénie (16 %), l’anémie (12 %) et la pneumonie (5 %) comparativement à la neutropénie (40 %) et la diarrhée (24 %) chez les sujets traités par l’association rituximab-idélalisib et à la neutropénie (31 %), l’anémie (9 %) et la constipation (6 %) chez ceux ayant reçu l’association rituximab-bendamustine.
Les taux d’hémorragie majeure ne se sont pas révélés supérieurs
Lors de l’essai ASCEND, les victimes de saignements ont été plus nombreuses parmi les patients traités par l’acalabrutinib que parmi les témoins (26 % vs 7,2 %), mais pas les victimes d’hémorragie majeure (1,9 % vs 2,6 %). En outre, les infections de grade 3 ou plus ont été moins répandues dans le groupe acalabrutinib que dans le groupe témoin (15 % vs 24 %).
La sélectivité relative des agents ciblés tels que l’acalabrutinib pourrait jouer un rôle important dans l’efficacité et l’innocuité de ces médicaments. Plusieurs autres inhibiteurs sélectifs de la BTK, dont le zanubrutinib et le loxo-305, en sont aussi au stade des programmes d’essais cliniques. Les premières données recueillies sur ces agents, qui ont été présentées lors de la réunion de 2019 de l’ASH, ont fait écho au principe voulant que la sélectivité à l’égard de la BTK peut améliorer la tolérabilité et l’efficacité du traitement opposé aux formes précoces ou avancées de la LLC.
« L’acalabrutinib est un inhibiteur hautement sélectif de la BTK qui forme une liaison covalente irréversible avec le site actif de cette dernière et qui fait preuve d’une activité minime envers les autres kinases », a déclaré le Dr Sharman. Il a ajouté que les réponses durables observées tant chez les patients jamais traités auparavant que chez ceux atteints d’une maladie récidivante ou réfractaire font de cet agent une nouvelle option thérapeutique très intéressante.
Conclusion
Les données collectées sur l’acalabrutinib pendant un essai de phase III mené chez des patients atteints de LLC jamais traitée auparavant ont été présentées dans le cadre de la réunion de 2019 de l’ASH, soit quelques semaines à peine après que la FDA ait autorisé l’utilisation de cet agent en pareille situation et dans les cas de LLC récidivante ou réfractaire. Pendant l’essai ELEVATE-TN, qui a été mené chez des patients atteints de LLC jamais traitée auparavant, la protection relative conférée contre l’évolution de la maladie ou le décès était plus forte que celle observée avec le même traitement standard que celui qui avait été utilisé dans le cadre des essais de phase III ayant porté sur l’ibrutinib, un inhibiteur de la BTK de première génération, ou sur le vénétoclax, un inhibiteur de la BCL-2. Lors de l’essai ASCEND, l’essai de phase III réalisé chez des patients atteints de LLC récidivante ou réfractaire, le traitement à prise orale a été mieux toléré et plus efficace que les traitements d’usage courant. L’ensemble des données ainsi collectées est venu étayer l’utilité d’une nouvelle option thérapeutique d’importance majeure pour le traitement de la LLC.