Neurologie
Conférence clinique et scientifique de 2021 de la Muscular Dystrophy Association (MDA)
Essai de phase III SPR1NT : une nouvelle thérapie génique permet de garder la maîtrise de l’amyotrophie spinale
Conférence en ligne – Les données actualisées de l’essai de phase III SPR1NT présentées dans le cadre de la conférence en ligne de 2021 de la MDA confirment qu’après une seule dose de cette thérapie génique homologuée depuis peu au Canada, la plupart des enfants atteints des pires formes d’amyotrophie spinale (AS) franchissent les étapes du développement propres à la petite enfance. Ces enfants ont été répartis en 2 cohortes selon qu’ils étaient porteurs de 2 ou 3 copies du gène SMN2. En juin 2020, date butoir fixée pour la collecte des données, la majorité des sujets des 2 cohortes continuaient d’afficher des gains soutenus.
Les effets positifs de cet agent, l’onasemnogène abéparvovec, sont remarquables puisque les enfants porteurs de 2 ou 3 copies du gène SMN2 ne survivent pas à la petite enfance s’ils ne sont pas traités. Selon le Dr Kevin A. Strauss, directeur médical de la Clinic for Special Children de Strasburg, en Pennsylvanie, beaucoup de nourrissons participant à cet essai de phase III ont franchi les étapes de développement correspondant à un âge déjà supérieur à leur espérance de vie.
Pour une entrevue exclusive avec le Dr Alex MacKenzie couvrant l’impact sur la pratique clinique, cliquez ici
Les sujets des deux cohortes ont été traités tôt
Les deux cohortes de l’essai SPR1NT ont fait l’objet de présentations distinctes à la conférence de la MDA. La première était composée de 14 patients porteurs de 2 copies du gène SMN2 et donc atteints de la forme la plus grave et la plus répandue d’AS. Selon le Dr Strauss, l’AS se définissant par des mutations du gène SMN1, le nombre de copies du gène SMN2 devient le meilleur facteur prévisionnel de la gravité de la maladie et du pronostic. À l’instar du gène SMN1, le gène SMN2 intervient dans la production de la protéine de survie du motoneurone (SMN, de l’anglais survival motor neuron) nécessaire à la maîtrise des symptômes.
« Une étude de l’évolution naturelle de l’AS chez des enfants non traités porteurs de 2 copies du gène SMN2 a révélé une survie médiane sans ventilation permanente de 10,5 mois. Par définition, ces patients sont incapables de s’asseoir sans aide », a précisé le Dr Krauss.
Le traitement a été amorcé dans les deux cohortes avant l’apparition des symptômes et dans les 6 semaines suivant la naissance, l’âge médian des sujets de la 1re cohorte étant alors de 20,6 jours. Les principaux critères d’exclusion étaient un faible poids (< 2 kg) et une atteinte des voies respiratoires mesurable d’après l’hypoxémie. Le paramètre d’évaluation principal était la capacité de s’asseoir sans aide pendant au moins 30 secondes. La survie, le recours nécessaire à une ventilation assistée et le score total à l’échelle III de Bayley pour la motricité fine et la motricité globale, étaient des paramètres d’évaluation secondaires ou exploratoires.
Étapes du développement franchies à un âge normal
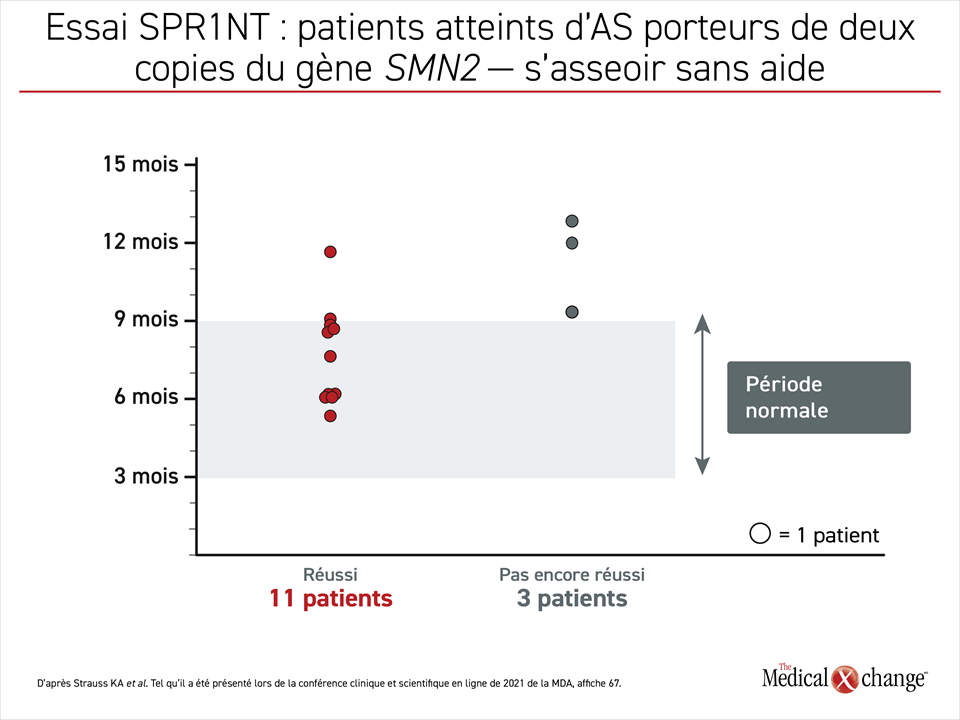
Selon les données collectées jusqu’à maintenant, 11 des (79 %) 14 patients de la 1re cohorte ont satisfait au paramètre d’évaluation principal en parvenant à s’asseoir sans aide (Figure 1). Tous sauf un y sont arrivés autour de l’âge normal de référence de 9,2 mois établi lors de l’étude multicentrique de l’OMS sur les valeurs de référence pour la croissance des enfants. Les 3 autres enfants n’ont pas encore franchi cette étape de leur croissance malgré leur âge.
« Tous les enfants sauf un qui ont satisfait au paramètre d’évaluation principal en parvenant à s’asseoir sans aide y sont arrivés autour de l’âge normal de référence. »
Plusieurs de ceux ayant satisfait au paramètre d’évaluation principal ont réussi à franchir les étapes de développement suivantes. En effet, 5 enfants peuvent se tenir debout sans aide et 3 d’entre eux y sont parvenus à l’âge de référence établi par l’OMS. Parmi les 5 enfants capables de se tenir debout tout seuls, 3 marchent sans aide. Encore là, ils y sont arrivés à un âge normal selon l’étude de l’OMS.
Beaucoup des patients de la 1re cohorte qui n’ont pas encore franchi les étapes plus avancées de leur développement sont encore à un âge où ils devraient normalement y parvenir.
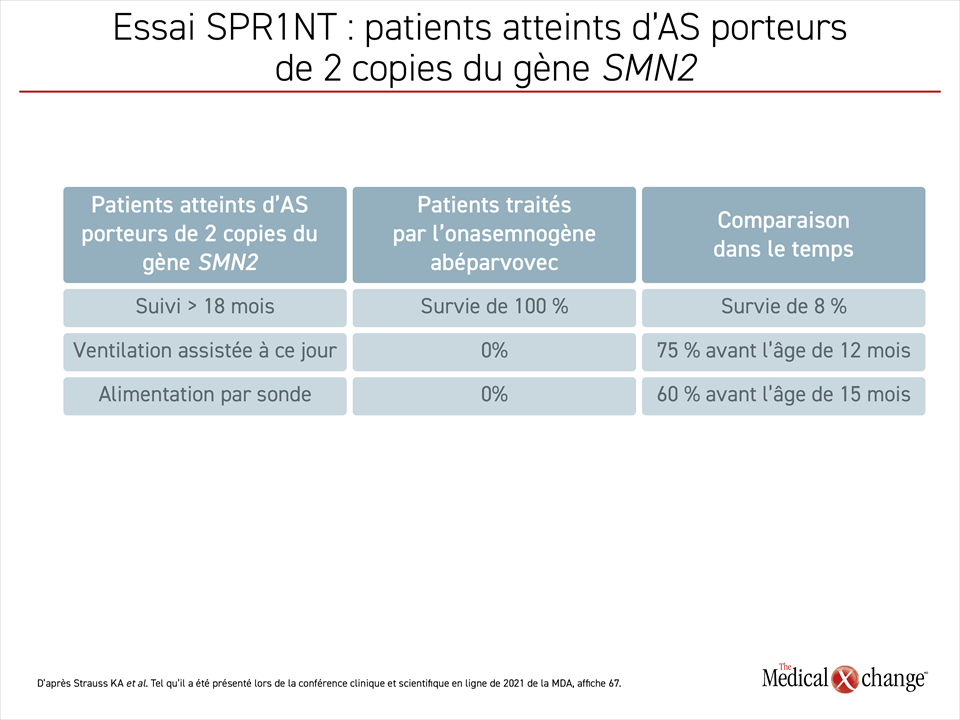
Les 14 patients ont continué d’acquérir des compétences, ce qui a été confirmé par les évaluations mensuelles réalisées au moyen l’échelle III de Bayley pour la motricité fine et la motricité globale réalisées jusqu’à la visite de contrôle la plus récente. Non seulement aucun d’eux n’a eu besoin de ventilation assistée, mais tous les résultats obtenus chez ces enfants, y compris leur survie, dépassent ceux observés chez des enfants atteints d’AS de type 1 ou 2 non traitée (Tableau 1).
Des paramètres d’évaluation plus stricts pour la 2e cohorte
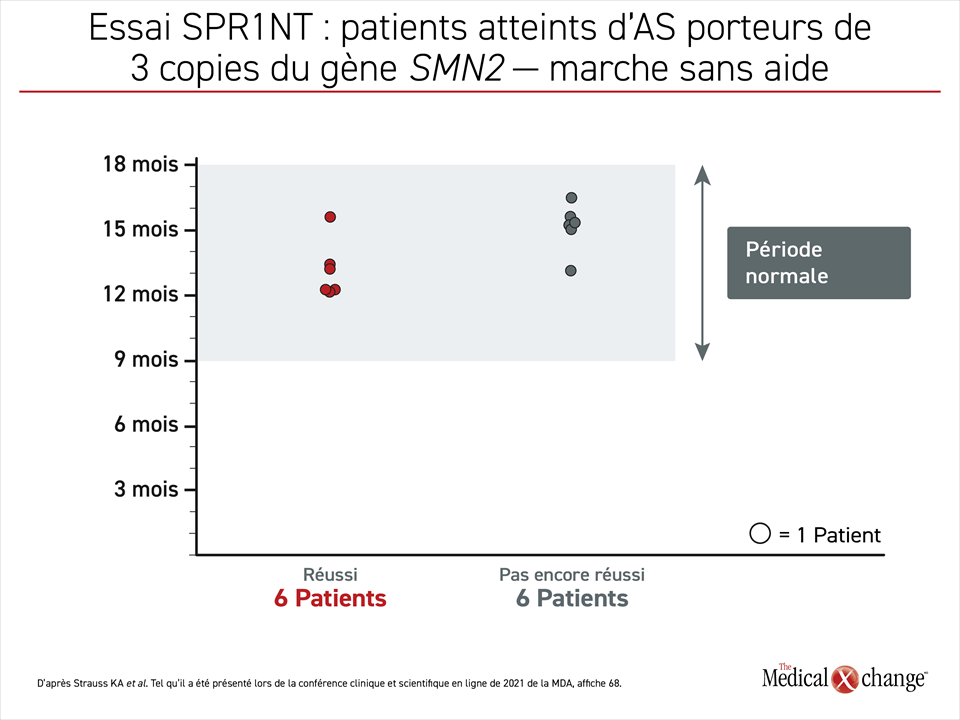
Les résultats enregistrés dans la 2e cohorte, qui était formée de 15 patients porteurs de 3 copies du gène SMN2, ont été tout aussi impressionnants. La capacité de se tenir debout pendant 3 secondes à au moins une des visites jusqu’à l’âge de 24 mois était le principal paramètre évalué. Les paramètres d’évaluation secondaires et exploratoires étaient les suivants : capacité de faire au moins 5 pas seul, aucune ventilation assistée jusqu’à l’âge de 24 mois et mesures objectives des habiletés motrices.
Sur les 15 patients de la 2e cohorte, 8 satisfont déjà au paramètre d’évaluation principal. Tous les autres sont encore à l’âge où ils devraient normalement y parvenir ou sont encore trop jeunes (Figure 2). Cela est aussi vrai pour les étapes de développement moins exigeantes telles que la capacité de s’asseoir sans aide, soit le principal paramètre évalué dans la 1re cohorte. Jusqu’à maintenant, 13 des 15 enfants de la 2e cohorte ont franchi cette étape et 8 réussissent à se tenir debout sans aide.
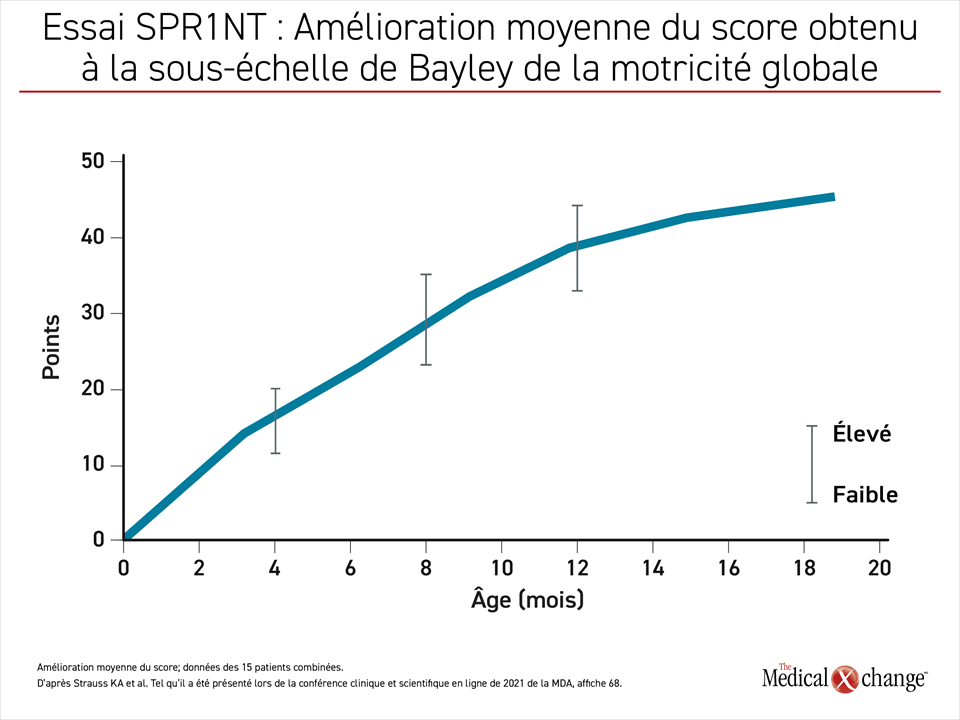
Comme ce fut le cas dans la 1re cohorte, l’amélioration mensuelle obtenue à la sous-échelle de Bayley de la motricité globale suit depuis le début du traitement une courbe ascendante constante qui n’a pas faibli jusqu’à maintenant (Figure 3).
Aucun des enfants n’a eu besoin de ventilation assistée jusqu’à maintenant
Comme ce fut le cas des enfants de la 1re cohorte, aucun de ceux de la 2e cohorte n’a eu besoin de ventilation assistée, d’une sonde d’alimentation entérale ni de quelque forme d’assistance vitale que ce soit, a rapporté le Dr Strauss.
Aucun problème grave d’innocuité n’a été soulevé dans l’une ou l’autre des cohortes de cet essai de phase III. Des effets indésirables possiblement liés au traitement ont été signalés chez 10 (71 %) et 7 sujets (47 %) des 1re et 2e cohortes respectivement, mais aucun n’était grave. Une hausse des enzymes hépatiques a été observée chez 29 % et 27 % des sujets de ces cohortes. Aucun cas d’incident cardiaque ou de thrombus n’a été signalé.
« Aucun problème grave d’innocuité n’a été soulevé dans l’une ou l’autre des cohortes de cet essai de phase III .»
Il faudra suivre ces patients plus longtemps pour confirmer qu’une seule dose d’onasemnogène abéparvovec leur permettra de continuer à franchir les étapes normales de leur développement. La période de suivi encore prévue à l’essai SPR1NT ne sera pas la seule à nous informer à ce chapitre. En effet, l’expérience acquise avec cet agent augmente grâce, entre autres, au registre RESTORE sur lequel repose une étude d’observation prospective multinationale visant à suivre des patients atteints d’AS pendant au moins 15 ans. Plus de 70 des patients qui sont inscrits au registre jusqu’à maintenant ont déjà reçu ou reçoivent de l’onasemnogène abéparvovec.
Ce n’est que récemment que des traitements opposés à l’AS ont pu être offerts
L’AS est une maladie neuromusculaire héréditaire rare contre laquelle il n’existait pas de traitement efficace, du moins jusqu’à dernièrement. Le premier médicament exerçant un effet direct sur l’AS, le nusinersen, a été homologué en 2017 au Canada. Cet agent, qui est injecté par voie intrathécale tous les 4 mois une fois que les doses d’attaque ont été administrées, modifie l’ARN messager de façon à augmenter les taux de protéine SMN produite par le gène SMN2. Le déficit en protéine SMN imputable aux mutations du gène SMN1 définissant l’AS est compensé par les médicaments qui amplifient la production de protéine SMN par le gène SMN2 même s’il est moins efficace à ce chapitre.
Récemment homologué au Canada et à l’étranger, l’onasemnogène abéparvovec est le deuxième médicament — et la première thérapie génique — qui soit opposé à l’AS. Une fois administré par voie intraveineuse (i.v.), l’onasemnogène abéparvovec, un vecteur de thérapie génique dérivé d’un virus adéno-associé (VAA), introduit un gène SMN1 fonctionnel dans les cellules transduites. Il a été mis au point pour stopper l’évolution de l’AS après une seule perfusion par voie i.v.
L’homologation du risdiplam, un médicament pour voie orale qui a fait preuve d’activité contre l’AS, est actuellement à l’examen. Tout comme le nusinersen, le risdiplam agit sur l’ARN messager de façon à amplifier la production de protéine SMN par le gène SMN2. Maintenant que des médicaments efficaces contre l’AS sont disponibles, plus d’attention sera accordée au dépistage et aux autres stratégies visant à diagnostiquer cette maladie rapidement. Le meilleur moyen d’améliorer l’issue de la maladie consiste probablement à amorcer le traitement avant même l’apparition des symptômes, surtout pendant la première enfance lorsque l’AS nuit à l’acquisition des habiletés motrices.
Les nouveaux traitements mettent en lumière l’importance d’un diagnostic précoce
Citant Linda P. Lowes, Ph. D., du Center for Gene Therapy du Nationwide Children’s Hospital de Columbus, en Ohio, qui était à la tête d’un essai de phase I sur l’onasemnogène abéparvovec, le Dr Strauss a déclaré : « L’importance de diagnostiquer et de traiter tôt la maladie pour améliorer davantage la fonction motrice avait déjà été mise en évidence ». Au cours de cet essai qui a servi à valider le bien-fondé de l’onasemnogène abéparvovec, la Dre Lowes a constaté que les enfants qui avaient entrepris le traitement avant l’âge de 3 mois ont franchi les étapes du développement plus tôt que ceux qui l’avaient amorcé plus tard, et ce indépendamment de la qualité de leur fonction motrice de départ (Lowes, L.P. et al. Pediatr Neurol. 2019;98:39-45).
Ces données et les résultats actualisés de l’essai SPR1NT « montrent bien l’importance de mettre sur pied des programmes de dépistage pour que les nouveau-nés qui ne montrent pas encore de symptômes soient traités rapidement », a affirmé le Dr Strauss.
Comme l’AS est une maladie rare dont l’incidence est estimée à 1 personne sur 100 000, il se peut qu’elle soit diagnostiquée tard, même dans les cas d’AS de type 1 dont les symptômes apparaissent quelques mois après la naissance. Selon certaines études, les retards de diagnostic frôlent les 4 mois en moyenne et peuvent dépasser 1 an dans les cas d’AS de type 2, ses symptômes se manifestant aussi pendant la première enfance (Lin, C.-W. et al. Pediatr Neurol 2015;53:293-300). L’espérance de vie théorique des patients atteints d’AS de type 2 est à peine plus longue que celle des patients aux prises avec l’AS de type 1, alors que dans les cas de types 3 et 4, caractérisés par un plus grand nombre de copies du gène SMN2, les patients peuvent espérer vivre jusqu’à l’âge adulte.
Les symptômes de l’AS de types 3 et 4 apparaissant parfois tard pendant l’enfance ou à l’âge adulte, le Dr Strauss considère que l’intérêt de diagnostiquer et de traiter tôt les cas de type 1 ou 2 réside dans la possibilité de permettre aux enfants d’acquérir des habiletés motrices normales et de les conserver. Il a précisé qu’une thérapie génique pouvant stabiliser les fonctions neurologiques en favorisant l’obtention de taux suffisants et soutenus de protéine SMN est un progrès majeur, gage de profonds changements dans le pronostic, même dans les formes les plus graves de la maladie.
Conclusion
Selon les données actualisées de l’essai de phase III SPR1NT, le lien entre l’amorce d’une nouvelle thérapie génique avant l’apparition des symptômes et une maîtrise soutenue de ces derniers chez les enfants porteurs de 2 ou 3 copies du gène SMN2 ne se dément pas. La plupart des sujets qui ont été suivis durant plus d’un an après avoir reçu une seule dose de ce médicament ont franchi les grandes étapes de leur développement et ont donc été capables de s’asseoir, de se tenir debout et de marcher, et ce à un âge normal pour beaucoup d’entre eux. Ce traitement a été bien toléré et n’a pas provoqué d’effets indésirables graves. Jusqu’à maintenant, aucun enfant n’a régressé après avoir franchi une étape de son développement et aucun n’est décédé. De plus, aucun enfant n’a eu besoin de ventilation assistée, d’une alimentation par voie entérale ni d’interventions visant à le garder en vie.