Neurology
Muscular Dystrophy Association (MDA) Clinical & Scientific Conference 2021
Spinal Muscular Atrophy Remains Controlled in a Phase 3 Study with New Gene Therapy: SPR1NT Trial
Virtual Meeting – After a single dose of a novel gene therapy recently approved in Canada, updated data from the phase 3 SPR1NT trial confirms that the majority of children with the most severe forms of spinal muscular dystrophy (SMA) are continuing to meet developmental milestones of early childhood. The trial, presented at the virtual 2021 MDA conference, evaluated this new gene therapy in two cohorts; those with two copies of SMN2 and those with three SMN2 copies. At the latest data cut taken in June 2020, sustained benefits continued to be observed in the majority of patients in both groups.
The benefits of this gene therapy, called onasemnogene abeparvovec, are remarkable because children with two or three SMN2 copies do not typically survive early childhood without treatment. In the phase 3 trial, many infants have now met expected developmental milestones at an age that has already exceeded their life expectancy, according to Dr. Kevin A. Strauss, Medical Director, Clinic for Special Children, Strasburg, Pennsylvania.
For an exclusive interview with Dr. Alex MacKenzie on the impact to clinical practice, click here
Both Study Cohorts Treat Early
Data from the two cohorts in the SPR1NT trial were presented separately at the MDA Conference. The first cohort was composed of 14 patients with two copies of the SMN2 gene, which is the most severe and common form. Due to mutations in the SMN1 gene, which defines SMA, the number of SMN2 copies is the best predictor of severity and prognosis, according to Dr. Strauss. Like SMN1, SMN2 mediates production of the survival motor neuron (SMN) protein needed for symptom control.
“In a natural history study of untreated patients with two copies of SMN2, the median survival without permanent ventilation was 10.5 months,” Dr. Krauss said. “By clinical definition, these patients do not sit without support.”
In both cohorts, treatment was initiated prior to the onset of symptoms and within six weeks of birth. The median age of treatment in cohort 1 was 20.6 days. The major exclusion criteria included low weight (<2 kg) and measurable airway compromise in the form of hypoxemia. The primary endpoint was the ability to sit without assistance for at least 30 seconds. Survival, need for ventilatory support, and Bayley-III fine and gross motor total score, were secondary or exploratory endpoints.
Milestones Achieved within Normal Development Window
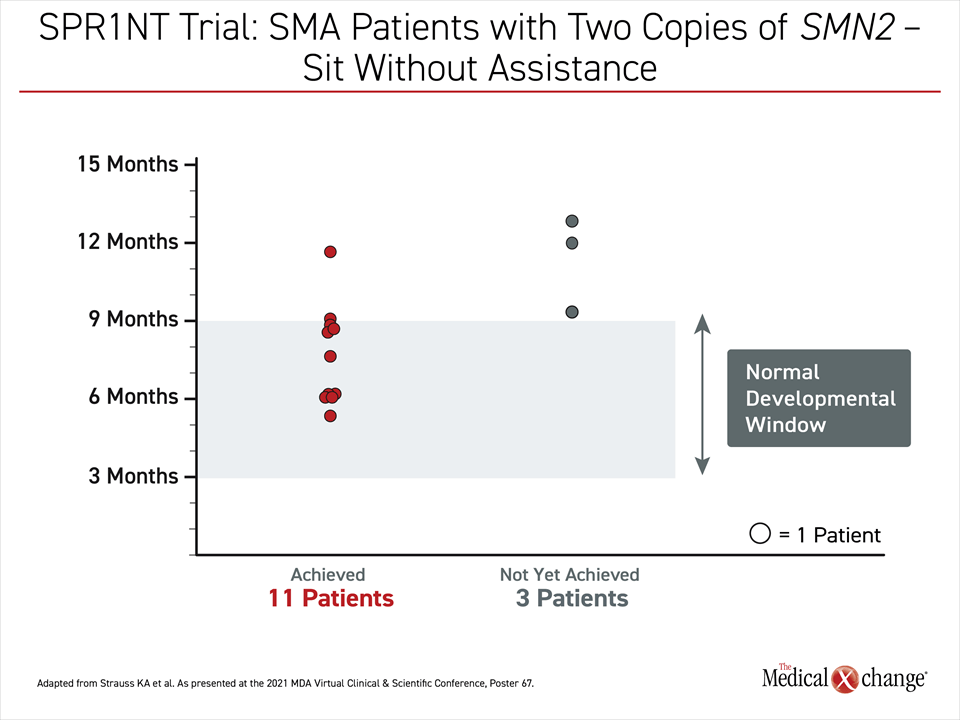
In follow-up so far, 11 (79%) of the 14 patients of cohort 1 achieved the primary endpoint of unassisted sitting (Figure 1). All but one patient achieved this milestone within the reference window of normal, which is 9.2 months, according to the World Health Organization Multicentre Growth Reference Study (WHO-MGRS). The three remaining patients have not yet met the age appropriate milestone.
“All but one of the patients who achieved the primary endpoint of sitting unassisted have done so within the reference window of normal development.”
Many of those who reached the primary endpoint have gone on to achieve subsequent developmental milestones. This includes five who are standing alone, of which three did so within the WHO-MGRS window of normal development. Of the five that achieved the standing milestone so far, three have also walked alone. All of these did so within the WHO-MGRS normal window for this milestone.
Of those in cohort 1 who have not yet met the more advanced milestones, many have not left the window of time in which these are expected in normal development.
When evaluated monthly with the Bayley-III subtests of gross and fine motor function, the accumulation of skills in all 14 patients has progressed steadily through the most recent follow-up. No patient has required ventilatory support. All of the outcomes in these children, including survival, surpass those observed in untreated children with type 1 or 2 SMA (Table 1).
More Rigorous Efficacy Endpoints for Cohort 2
In the second cohort, results have been equally impressive. In this cohort, consisting of 15 patients with three copies of SMN2, the primary endpoint was ability to stand for at least three seconds at any visit up to 24 months. Secondary and exploratory endpoints included independent walking for at least five steps, freedom from respiratory support up to age 24 months and objective measures of motor skills.
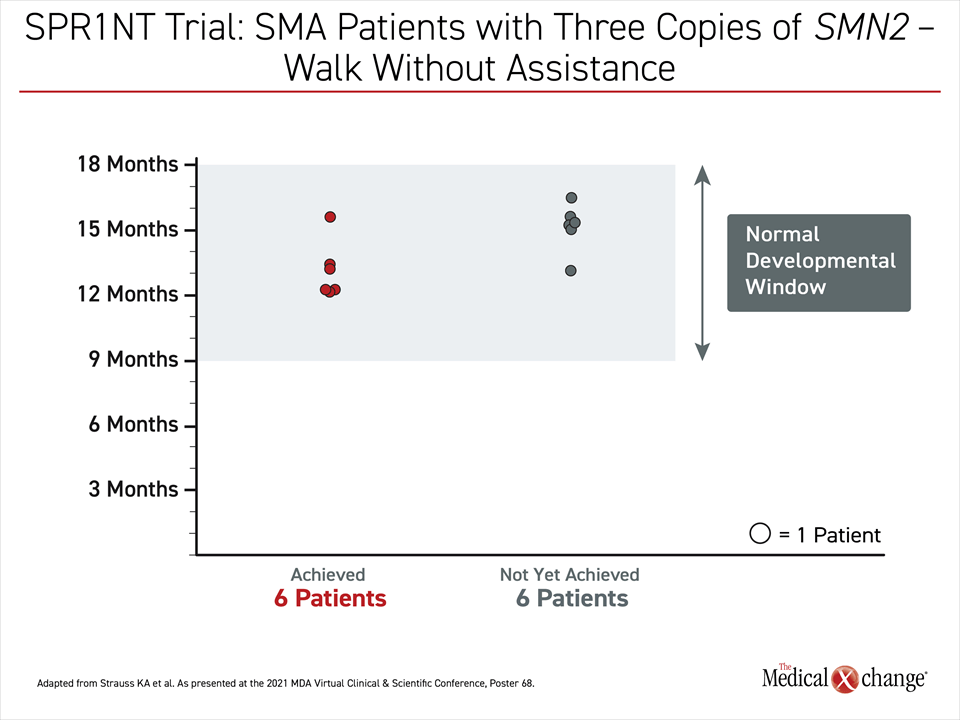
Of the 15 patients in cohort 2, eight have already reached the primary endpoint. All of the remaining patients remain within or have not reached the time window in which this milestone of development is typically reached (Figure 2). This is also true of less rigorous milestones, such as sitting without assistance, which was the primary outcome defined for cohort 1. So far, this milestone has been achieved by 13 of 15 infants in cohort 2. Standing without assistance has been achieved so far by eight of 15 children.
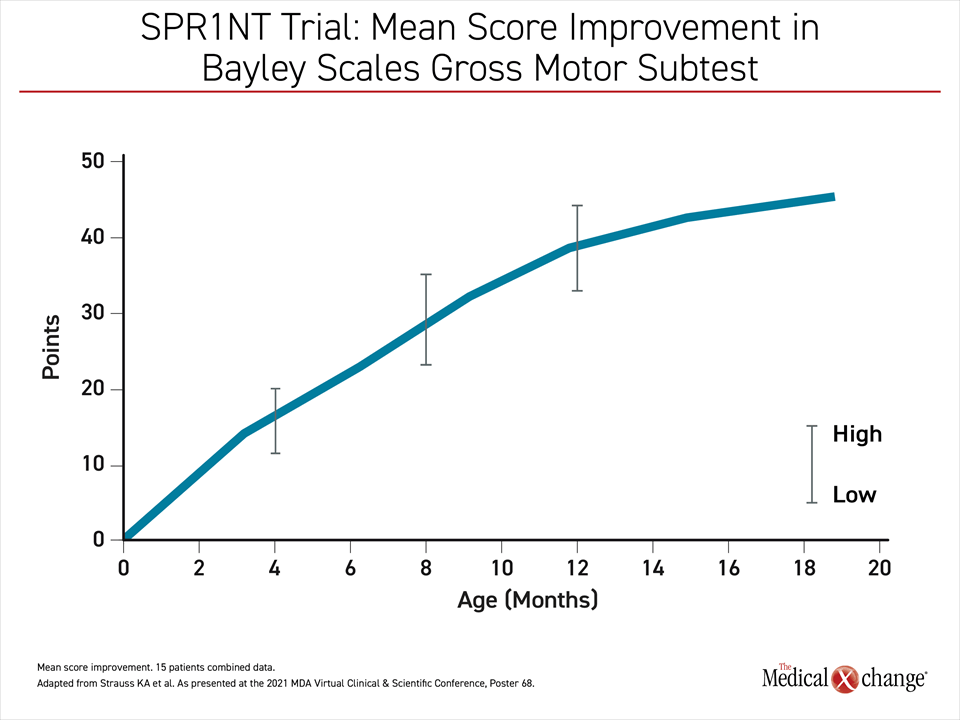
As in cohort 1, the monthly improvement in the Bayley Scales gross motor subtest has followed a steady upwards progression that began with initiation of therapy through follow-up so far (Figure 3).
No Ventilatory Support Required So Far
As in cohort 1, no patient in cohort 2 has required ventilatory support, gastric feeding tube, or any other form of life support, Dr. Strauss reported.
There have been no serious safety issues observed in either cohort of this phase 3 trial. Adverse events potentially related to treatment were seen in 10 (71%) of those in cohort 1 and seven (47%) in cohort 2, but none were serious. Liver enzyme elevations were observed in 29% and 27% of these two cohorts, respectively. There have been no reports of cardiac events or thrombi.
“There have been no serious safety issues observed in either cohort of this phase 3 trial.”
Longer follow-up is needed to confirm that a single dose of onasemnogene abeparvovec will continue to permit acquisition of normal developmental milestones indefinitely. In addition to further planned follow-up of the SPR1NT trial, experience with onasemnogene abeparvovec is accruing from additional sources, such as the RESTORE registry. This prospective multinational observational study is designed to follow SMA patients for at least 15 years. Of the patients entered into the registry so far, more than 70 patients have received or are receiving onasemnogene abeparvovec.
SMA Therapies Are Only Recently Available
SMA is a rare inherited neuromuscular disorder. Until recently, there were no effective treatments. The first medication with a direct impact on SMA, nusinersen, was approved in Canada in 2017. This therapy, which is administered via intrathecal injection every four months after a loading dose regimen is completed, modifies messenger RNA to enhance SMN levels generated by the SMN2 gene. The deficiency in SMN caused by the SMN1 mutations that define SMA are compensated by treatments that increase SMN2-mediated production of SMN even though the SMN2 gene is less efficient for SMN production.
When it was recently approved in Canada and elsewhere, onasemnogene abeparvovec became the second treatment and the first gene therapy for SMA. When delivered through an adeno-associated viral vector intravenously (IV), onasemnogene abeparvovec provides a functional SMN1 gene. This therapy was developed to halt SMA progression after a single IV infusion.
Approval for a third therapy, called risdiplam, which has shown activity when administered orally, is now being sought in Canada. Like nusinersen, risdiplam modifies mRNA activity to increase production of SMN from the SMN2 gene. With effective treatment options for SMA, more attention will be paid to screening and other strategies for early diagnosis. The greatest opportunity to improve outcomes is likely to be gained from initiating treatments in advance of symptoms, particularly in infancy when SMA threatens the ability to gain motor skills.
New Therapies Stress Need for Early Diagnosis
“The importance of early diagnosis and treatment to achieve greater improvements in motor function has been stressed previously,” reported Dr. Strauss, citing a phase 1 trial with onasemnogene abeparvovec led by Linda P. Lowes, PhD, Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, Ohio. In that phase 1 study, which provided a proof of concept for onasemnogene abeparvovec, Dr. Lowes showed that those initiated on therapy prior to three months of age achieved milestones earlier than those initiated later regardless of baseline motor function. (Lowes LP et al. Pediatr Neurol. 2019;98:39-45).
These data along with the current results of the SPR1NT trial “underscore the importance of implementation of newborn screening programs to facilitate early treatment administration in presymptomatic patients,” Dr. Strauss said.
“These data ‘underscore the importance of implementation of newborn screening programs to facilitate early treatment administration.’”
Due to the rarity of SMA, for which the estimated incidence is one per 100,000 individuals, the diagnosis can be delayed even in type 1 disease, which becomes symptomatic within months of birth. Studies have shown that diagnostic delay averages nearly four months and can exceed a year in type 2 SMA, which also typically becomes symptomatic in early infancy (Lin C-W et al. Pediatr Neurol. 2015;53:293-300). The expected lifespan of type 2 SMA, unlike type 3 or type 4 SMA, which are associated with more copies of the SMN2 gene and often permit survival into adulthood, is only modestly greater than that of type 1.
While the symptoms of types 3 and 4 SMA might not develop until late childhood or adulthood, the value of early diagnosis and early therapy for types 1 or 2 SMA is the potential to develop and preserve normal acquisition of motor skills, according to Dr. Strauss. Ultimately, he suggested that the introduction of a gene therapy that yields early and sustained production of adequate levels of SMN to stabilize neurologic function is a major advance. The promise is a profound change in prognosis even among those with the most severe forms.
Conclusion
Updated data from the phase 3 SPR1NT trial continues to associate presymptomatic initiation of a new gene therapy with sustained symptom control in infants with either 2 or 3 copies of SMN2. For those followed for more than one year after a single dose of this therapy, most have achieved major developmental milestones, such as sitting, standing, and walking. In many patients, these milestones are achieved within normal developmental windows. The therapy has been well tolerated with no serious adverse events. To date, no milestones once achieved have been lost and all patients are alive. No patient so far has required ventilatory support, feeding tubes, or other interventions required to ensure survival.