Neurologie
Réunion annuelle de 2021 de l’American Academy of Neurology (AAN)
Pour être efficaces, les traitements dirigés contre l’amyotrophie spinale doivent être entrepris tôt
Réunion en ligne – Qu’il soit utilisé comme monothérapie initiale ou en remplacement d’un autre agent, le vecteur de thérapie génique axé sur le gène codant pour la protéine de survie du motoneurone 1 (SMN1) offrirait une amélioration plus marquée et plus rapide des capacités fonctionnelles qu’un traitement ciblant le gène SMN2 aux jeunes enfants traités contre une amyotrophie spinale (AS) après l’âge de 6 mois. Les résultats ont été évalués pendant le laps de temps où ces deux traitements sont indiqués, ce qui peut être significatif pour le pronostic à long terme. Les données actualisées sur la thérapie génique administrée en contexte présymptomatique confirment l’obtention d’une réponse soutenue pendant une période de suivi qui s’étend maintenant sur 5,6 années.
Le nombre de médicaments contre l’AS homologués au Canada est passé de 0 à 3 au cours des 3 dernières années. Deux d’entre eux reposent sur une stratégie élémentaire. Le nusinersen et le risdiplam, le tout dernier à avoir été homologué, agissent sur l’activité du gène SMN2 de façon à amplifier la production de protéine SMN dans le cadre d’un traitement d’entretien. En remplaçant le gène SMN1 manquant, l’onasemnogène abéparvovec, le vecteur de thérapie génique, a permis une production soutenue de protéine SMN après une seule dose. Bien que cela reste à prouver, la production de protéine SMN pourrait être assurée la vie durant.
L’onasemnogène abéparvovec est homologué chez les enfants âgés de 24 mois ou moins, mais il a surtout été étudié chez des enfants de moins de 6 mois. Le nusinersen et le risdiplam sont homologués chez les enfants et les adultes. Comme aucun essai comparatif n’a été réalisé, on ne connaît pas l’efficacité relative de ces agents chez les enfants âgés de 6 à 24 mois pour lesquels les deux classes de médicaments sont indiquées.
Comparaison des traitements opposés à l’AS chez les enfants âgés de 6 à 24 mois
« Les traitements de fond ont énormément amélioré le pronostic des enfants atteints d’AS, mais nous disposons de peu de données nous permettant de tenir compte de l’efficacité relative de ces traitements chez les enfants de plus de 6 mois, qu’ils suivent toujours leur traitement initial ou qu’il ait été remplacé par un autre », a déclaré le Dr Neil Minkoff, directeur de FountainHead HealthCare, de Sudbury, au Massachusetts, et coauteur d’une étude visant à obtenir des données sur la pratique clinique grâce à un sondage auprès de médecins.
Vingt-deux médecins ayant déjà traité l’AS, le trouble neuromusculaire rare causé par la délétion ou la mutation des deux copies du gène SMN1, ont répondu à ce sondage mené avant l’homologation du risdiplam au Canada. Ils y ont rapporté les résultats obtenus chez 30 enfants âgés de 6 à 24 mois ayant reçu de l’onasemnogène abéparvovec seulement, chez 54 enfants recevant du nusinersen depuis le début de leur traitement et chez 19 enfants ayant entrepris leur traitement par le nusinersen, mais qui sont passés à l’onasemnogène abéparvovec par la suite.
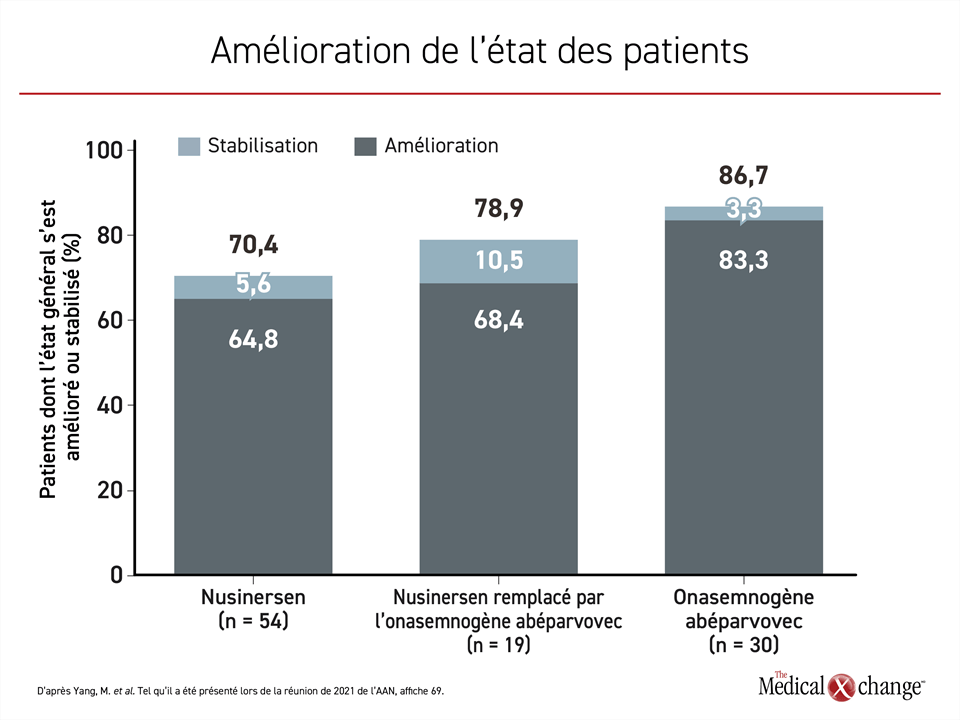
L’état des enfants des 3 groupes s’est amélioré, mais l’amélioration était plus marquée chez ceux traités par l’onasemnogène abéparvovec, un agent qui introduit une copie entièrement fonctionnelle du gène SMN1 dans les motoneurones afin de rétablir la production de protéine SMN. Par rapport au début du traitement, la proportion des enfants dont l’état s’est amélioré s’élevait à 83,3 % et celle des enfants dont l’état s’est stabilisé, à 3,3 %. L’état de 64,8 % des enfants traités par le nusinersen s’est amélioré et il s’est stabilisé chez 5,6 % des enfants. Le nusinersen est un oligonucléotide antisens qui, à l’instar du risdiplam, un agent plus récent, amplifie la production de protéine SMN en ciblant le gène SMN2. Quant aux enfants passés du nusinersen à l’onasemnogène abéparvovec, 68,4 % ont pris du mieux et 10,5 % ont vu leur état se stabiliser (Figure 1).
Amélioration des capacités fonctionnelles grâce à la thérapie génique administrée en première intention
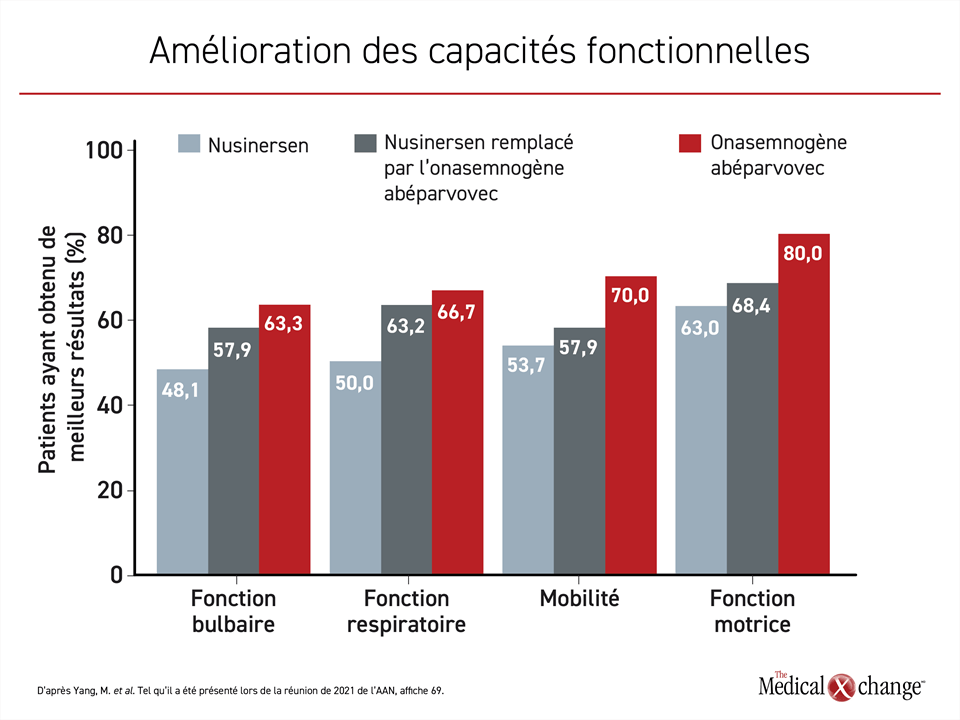
La comparaison de ces trois mêmes groupes a permis de constater que l’amélioration de certaines capacités fonctionnelles suivait la même gradation. Il y avait un avantage pour la mobilité et les fonctions motrice, bulbaire et respiratoire à commencer le traitement avec de l’onasemnogène abéparvovec plutôt qu’avec du nusinersen en monothérapie ou à le remplacer par l’onasemnogène abéparvovec (Figure 2).
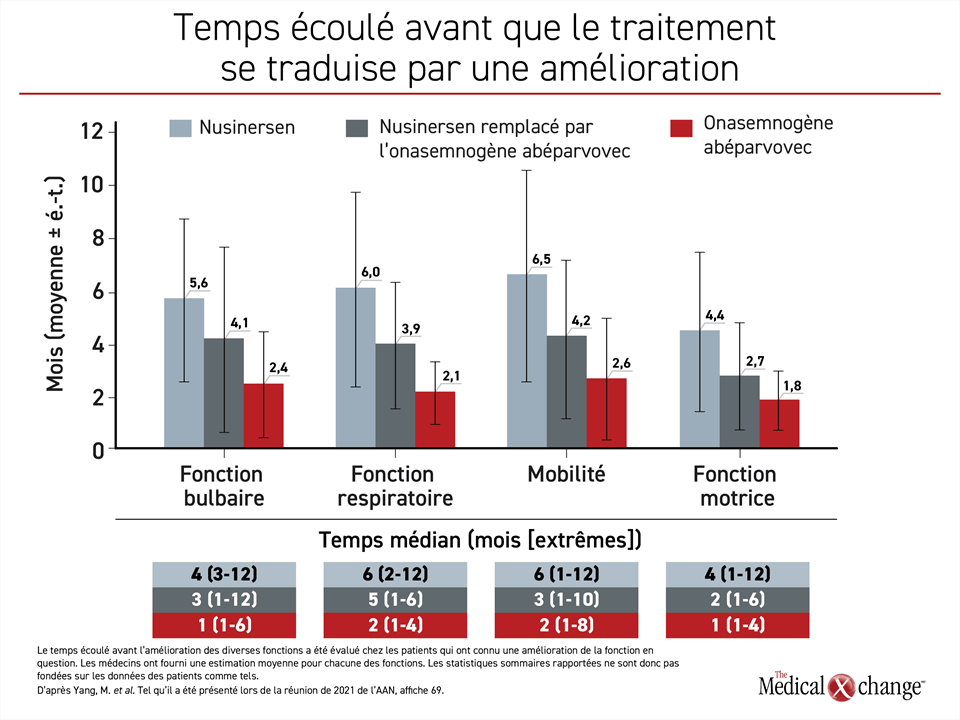
Il a aussi fallu moins de temps aux patients recevant la thérapie génique ciblant le gène SMN1 pour que leurs capacités fonctionnelles s’améliorent. Les patients passés du nusinersen à l’onasemnogène abéparvovec ont mis 2 mois de moins environ pour connaître une telle amélioration que ceux ayant pris du nusinersen seulement. Quant à ceux traités d’emblée par l’onasemnogène abéparvovec, ce délai a été écourté de 2 mois supplémentaires environ (Figure 3). Comme l’AS risque d’entraîner une perte irréversible de capacités fonctionnelles au cours de son évolution, une réponse obtenue plus tôt et mieux préservée est probablement bénéfique à long terme.
La gravité de l’AS est une variable importante pour qui veut analyser la réponse et le sort des patients à long terme. Dans l’AS de type I, qui représente environ 60 % des cas, la perte de fonction du gène SMN1 est liée à une espérance de vie théorique de moins de 2 ans. Les symptômes apparaissent habituellement plus tardivement chez les patients atteints d’AS de type II ou plus, souvent après qu’ils aient franchi des étapes importantes de leur développement. Dans les cas d’AS de type III, plus rares, il se peut que les symptômes ne se manifestent pas avant l’adolescence ou même l’âge adulte, ce qui est en grande partie imputable au nombre de copies du gène SMN2 qui produit moins de protéine SMN que le gène SMN1, quoiqu’en quantité suffisante pour retarder l’apparition des symptômes et en atténuer la gravité.
Le développement neurologique pourrait exiger un traitement précoce
Il est essentiel de diagnostiquer et de traiter l’AS tôt, surtout celle de type I, pour permettre aux enfants de franchir normalement les étapes de leur développement. Lors des études sur l’onasemnogène abéparvovec, des enfants atteints d’AS de type I ont entrepris leur traitement peu de temps après l’apparition des symptômes et dans certains cas, avant. Selon une mise à jour des programmes d’études cliniques LT-001 et LT-002 sur cet agent, les résultats obtenus ont été remarquables.
Dans le cadre du programme LT-001, qui comprend les données initiales de la première phase de l’essai START, l’âge moyen des 12 patients ayant reçu une dose thérapeutique est de 5,2 ans. L’aîné est maintenant âgé de 6,1 ans, selon le Dr Jerry R. Mendell, président de la recherche en pédiatrie, au Nationwide Children’s Hospital, de Columbus, en Ohio.
« [Chez les patients de 6,1 ans ou moins,] nous assistions à une persistance prolongée de la fonction génique. »
« Nous assistions à une persistance prolongée de la fonction génique », a-t-il affirmé. Dans ce groupe d’enfants pour lesquels l’espérance de vie se calcule en mois s’ils ne sont pas traités, « aucun n’est sous ventilation permanente ».
Le suivi de longue durée est rassurant
Le suivi réalisé dans le cadre des études du programme LT-002, dont certaines ont mené à l’homologation du produit, dure maintenant depuis 2,7 ans. Le traitement des 22 patients symptomatiques qui ont participé à l’étude STR1VE a été entrepris alors qu’ils étaient âgés de 3,7 mois en moyenne. La plupart d’entre eux ont franchi les étapes de leur développement à un âge jugé normal. Pendant ce suivi, notamment celui des patients recevant des doses thérapeutiques dans le cadre du programme LT-001, aucun d’eux n’a eu besoin de ventilation assistée prolongée, d’une sonde d’alimentation entérale ni de quelque forme d’assistance vitale que ce soit.
Les données sur l’innocuité sont rassurantes aussi. D’après le Dr Mendell : « Pendant le suivi de longue durée des programmes LT‑001 et LT-002, il n’y a eu aucun nouveau signal d’alarme évoquant un problème d’innocuité ».
Toutes les études menées dans le cadre du programme LT-002 sont en cours et il est prévu que le suivi dure encore plusieurs années. Alors que l’étude STR1VE porte sur l’administration de la thérapie génique à des enfants symptomatiques, l’étude SPR1NT sert à évaluer l’amorce du traitement avant l’apparition des symptômes. Toutes ces études sont assorties d’une période de suivi suffisamment longue pour fournir des données probantes solides sur l’efficacité et l’innocuité du traitement (Figure 4).
Compte tenu de l’importance de maintenir des taux suffisants de protéine SMN, il y a un avantage à administrer une dose unique de thérapie substitution génique plutôt que d’administrer des doses d’entretien à vie. Même si le nusinersen, le premier agent homologué contre l’AS, a marqué une percée décisive dans la maîtrise de l’AS, de nouvelles données sur l’observance du traitement en pratique clinique, qui repose sur des injections intrathécales administrées tous les 4 mois une fois que les doses d’attaque ont été données, indiquent qu’il est fréquent que des doses soient omises, ce qui a des conséquences sur l’atteinte et la pérennité des étapes du développement.
Évaluation de l’adhésion et de la persévérance durant le traitement d’entretien
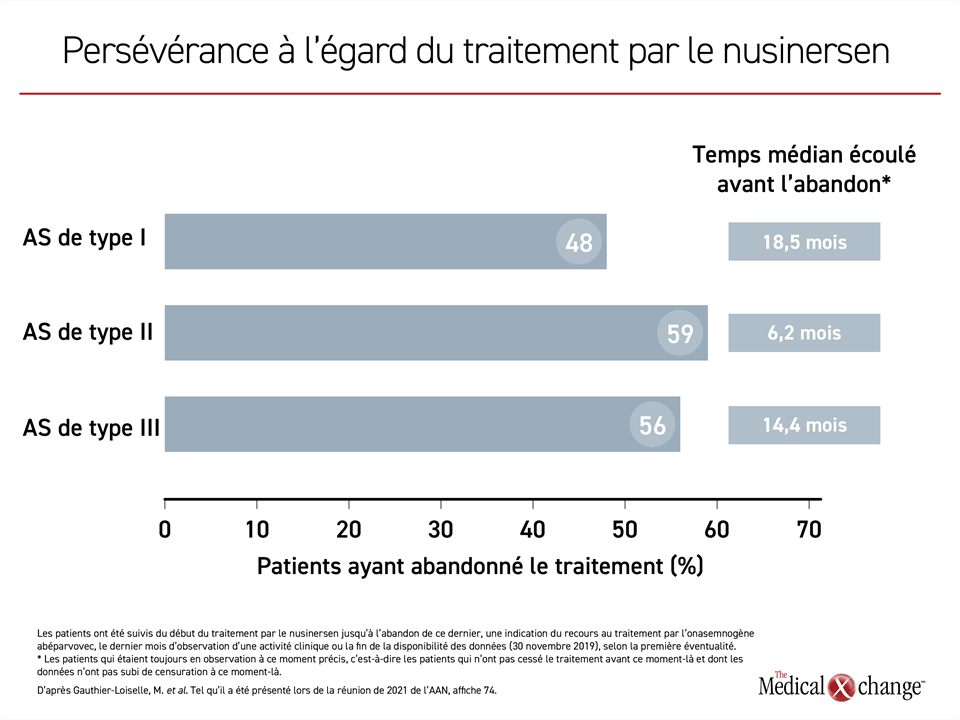
Le respect du traitement chez des patients atteints d’AS de type I, II ou III ayant terminé la phase d’attaque du nusinersen et qui étaient inscrits dans une base de données étatsunienne a été évalué sur une période d’observation suffisamment longue pour qu’on puisse présumer qu’ils avaient reçu au moins 4 doses d’entretien conformément aux indications de la monographie. Cette évaluation a porté sur 23 patients atteints d’AS de type I, 41 patients aux prises avec le type II de la maladie et 260 patients affligés du type III.
La proportion de patients ayant reçu au moins une dose décalée, c’est-à-dire espacée de plus de 120 jours de la précédente, était considérable. Elle se chiffrait à 52,2 %, à 70,7 % et à 53,1 % dans les cas d’AS de types I, II et III, respectivement. La proportion d’abandons pendant la période de suivi était également importante, ce qui a, semble-t-il, eu un effet sur le sort des patients (Figure 5).
« Indépendamment du type d’AS dont ils étaient atteints, les patients n’ayant pas respecté le traitement risquaient davantage de présenter des affections concomitantes associées à l’AS, dont des problèmes d’alimentation, de la dyspnée et d’autres troubles respiratoires, un retard de croissance et une faiblesse musculaire », a affirmé la Dre Marjolaine Gauthier-Loiselle, gestionnaire chez Analysis Group, de Montréal, au Québec.
« Indépendamment du type d’AS dont ils étaient atteints, les patients n’ayant pas respecté le traitement risquaient davantage de présenter des affections concomitantes associées à l’AS, dont des problèmes d’alimentation. »
Il a été impossible d’expliquer le manque d’adhésion au traitement par le nusinersen à partir des données tirées de la pratique clinique, mais les chercheurs ont émis l’hypothèse que des obstacles logistiques, tels que les déplacements vers les établissements où sont administrées les nombreuses doses d’entretien, pourraient figurer parmi les explications possibles. La douleur causée par les injections intrathécales trimestrielles pourrait en être une autre. Cette analyse rétrospective n’a pas permis d’établir un lien causal entre le manque d’adhésion au traitement et une hausse du risque d’affections concomitantes, mais il est logique de supposer que les retards de traitement et les abandons « se soldent par une perte d’habiletés motrices et l’évolution de la maladie », a déclaré la Dre Gauthier-Loiselle.
La thérapie génique chez les patients porteurs de plus de 2 copies du gène SMN2
La thérapie génique est particulièrement intéressante pour les patients atteints d’AS qui sont porteurs de deux copies du gène SMN2 seulement étant donné que les traitements ciblant ce gène ne peuvent stimuler qu’une production restreinte de protéine SMN. Pour ce qui est des préoccupations suscitées par l’adhésion au traitement, l’efficacité de la thérapie génique chez les patients porteurs de plus de 2 copies du gène SMN2 peut toutefois être utile à ce chapitre. Selon les données recueillies chez les 15 patients porteurs de 3 copies du gène SMN2 ayant participé à l’étude SPR1NT, la survie sans incident s’élève à 100 % après un suivi allant jusqu’à 21 mois.
« Treize des 15 enfants de la cohorte des porteurs de 3 copies étaient capables de s’asseoir seuls. Ils ont presque tous franchi cette étape du développement à un âge jugé normal », a rapporté le Dr Kevin A. Strauss, directeur médical de la Clinic for Special Children de Strasburg, en Pennsylvanie. Les 2 enfants qui n’y sont toujours pas parvenus sont encore à un âge où ils devraient normalement y arriver.
« Treize des 15 enfants de la cohorte des porteurs de 3 copies réussissaient à s’asseoir seuls. Ils y sont presque tous arrivés autour de l’âge normal de référence établi pour cette étape du développement. »
Huit des 15 enfants peuvent se tenir debout et 6 sont capables de marcher sans aide. Aucun des enfants qui ne réussissent pas encore à le faire n’a dépassé l’âge où il devrait normalement franchir ces étapes et certains autres sont encore trop jeunes.
« Lors de leur dernière visite, les 15 enfants porteurs de 3 copies du gène SMN2 ont affiché sur l’échelle III de Bayley une performance motrice finale comparable à celle des enfants en santé appariés en fonction de l’âge », a affirmé le Dr Strauss.
Conclusion
Il existe aujourd’hui trois options de traitement contre l’AS dotées de mode d’action différent. Contrairement aux agents exigeant un traitement d’entretien comme le nusinersen et le risdiplam, dont l’homologation est récente, une seule perfusion d’onasemnogène abéparvovec suffit pour rétablir l’activité du gène SMN1. Ces médicaments n’ont pas fait l’objet d’une comparaison directe, mais les médecins expérimentés ayant participé au sondage ont affirmé que l’amélioration des habiletés motrices et la régression des déficits associés à l’AS sont survenues plus rapidement quand le traitement était amorcé avec de l’onasemnogène abéparvovec plutôt qu’avec du nusinersen. Les patients ayant remplacé le nusinersen par de l’onasemnogène abéparvovec ont acquis des habiletés motrices et franchi les grandes étapes du développement plus tôt que ceux ayant poursuivi leur traitement par le nusinersen. À terme, l’administration de l’onasemnogène abéparvovec en une dose unique élimine les problèmes d’observance. Les enfants qui ont participé aux premiers essais sur l’onasemnogène abéparvovec sont suivis depuis plus de 5 ans maintenant. Or les effets positifs, l’innocuité favorable et la tolérabilité de cet agent sont toujours au rendez-vous.