Neurology
American Academy of Neurology (AAN) Annual Meeting 2021
Efficacy of Spinal Muscular Atrophy Therapies Depends on Early Initiation
Virtual Meeting – In young children treated for spinal muscular atrophy (SMA) after the age of six months, the vector-based survival motor neuron-1 (SMN1) gene therapy appears to provide greater and faster functional improvement than a treatment targeted at the SMN2 gene when used either as initial monotherapy or switched over. The outcomes were evaluated during the window of time in which both drugs have an indication. This is likely relevant to long-term prognosis. In newly-updated data with gene therapy administered presymptomatically, sustained response has been observed with gene therapy in follow-up now extending to 5.6 years.
The number of SMA therapies approved in Canada has increased from zero to three in the past three years. Of these three treatments, there are two basic strategies. Nusinersen and the most recently approved therapy, risdiplam, work by modifying SMN2 gene activity to increase SMN protein production through maintenance dosing. By replacing the missing SMN1 gene, the vector-based SMN1 gene replacement therapy onasemnogene abeparvovec has provided sustained SMN production after a single dose. Although not yet proven, the potential is for lifetime SMN protein production.
Onasemnogene abeparvovec is approved in children up to 24 months of age but has been studied primarily in children treated before the age of six months. Nusinersen and risdiplam are approved in children and adults. There are no comparative trials, so there has been a knowledge gap concerning relative efficacy in children six to 24 months of age, where both classes of drugs are indicated.
SMA Therapies Compared in Children between 6-24 Months
“All of the disease-modifying therapies have dramatically improved the prognoses of children with SMA, but there are limited data with which to consider the relative efficacy of treatments among children over the age of six months—whether they have remained on initial treatment or switched from another therapy,” reported Dr. Neil Minkoff, Director, FountainHead HealthCare, Sudbury, Massachusetts. Dr. Minkoff was among coauthors of a study intended to derive real-world data from a physician survey.
The survey collected responses from 22 providers who have treated SMA, the rare genetic neuromuscular disorder caused by deletion or mutation of both copies of the SMN1 gene. Conducted before risdiplam was licensed in Canada, the responding providers reported outcomes in 30 children between the ages of 6 and 24 months who were started and remained on onasemnogene abeparvovec, 54 children who started and remained on nusinersen, and 19 children who started on nusinersen and were then switched to onasemnogene abeparvovec.
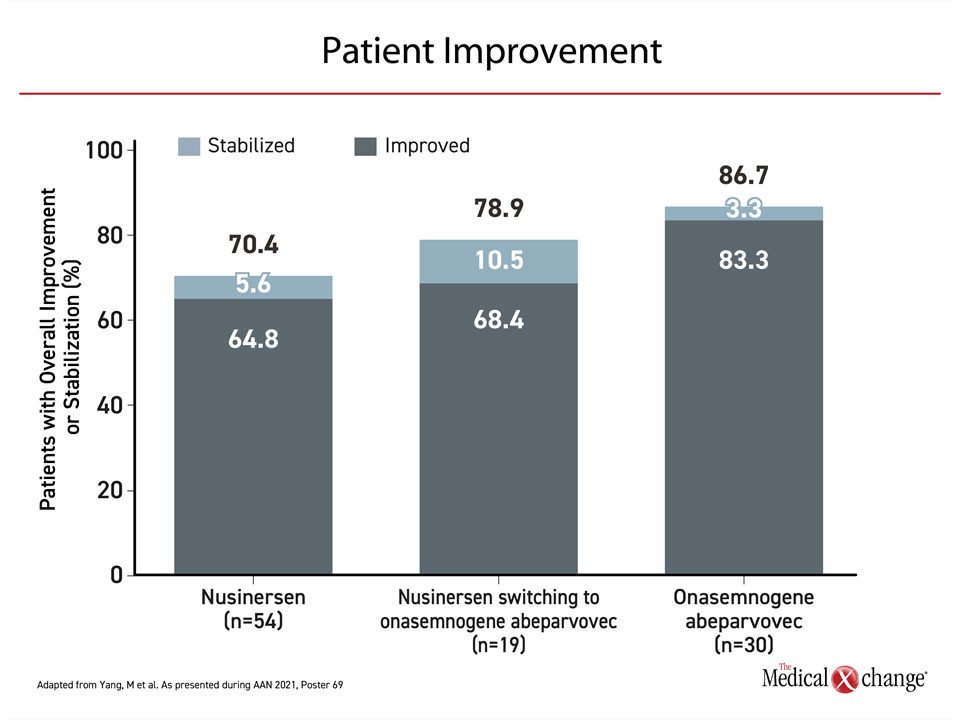
Improvement was demonstrated in all three groups but the relative improvement was greater among those who received onasemnogene abeparvovec, which delivers a fully functional copy of the SMN gene into motor neurons to restore SMN protein production. Relative to baseline, 83.3% improved and an additional 3.3% were stabilized. On nusinersen, an antisense oligonucleotide that, like the newer risdiplam, increases SMN protein production by targeting the SMN2 gene, improvement was achieved in 64.8% with an additional 5.6% stabilized. For those initiated on nusinersen and switched to onasemnogene abeparvovec, 68.4% improved with an additional 10.5% stabilized (Figure 1).
Functional Improvement with First-line Gene Therapy
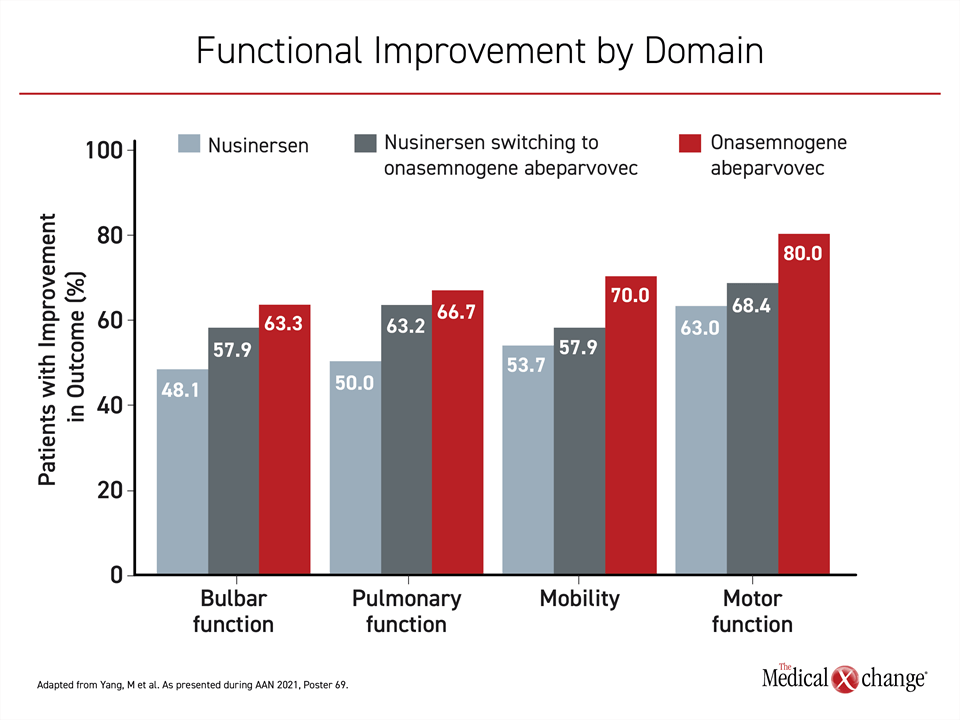
The same stepwise relationship was observed on specific functional improvements when the same three groups were compared. For motor function, mobility, bulbar function, and pulmonary function, there was a reported advantage to starting patients with onasemnogene abeparvovec vs nusinersen monotherapy and also switching to onasemnogene abeparvovec from nusinersen (Figure 2).
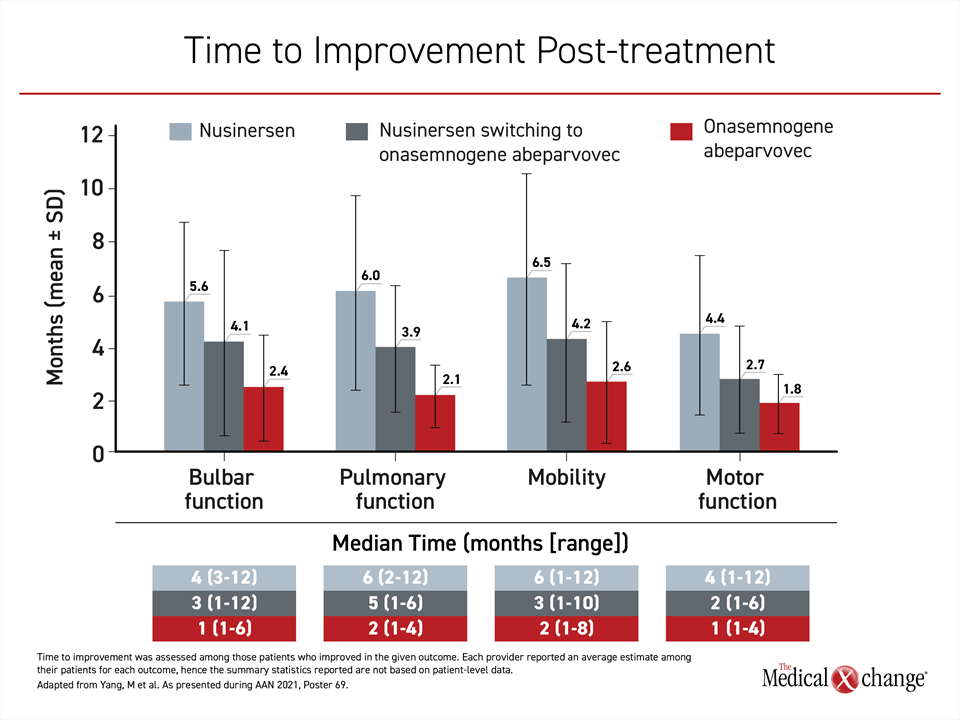
The time to these functional improvements was also shorter in patients receiving SMN1 targeted gene therapy. For those switched from nusinersen to onasemnogene abeparvovec, the time to improvement was reduced by approximately two months relative to those on nusinersen monotherapy. For those initiated on onasemnogene abeparvovec, there was a further reduction of approximately two months (Figure 3). Due to the risk for irreversible functional loss from SMA progression, the earlier response and greater maintenance of response is likely relevant to long-term prognosis.
In patients with SMA, disease severity is an important variable in analyzing response and long-term outcomes. In type 1 SMA, which accounts for approximately 60% of cases, the lost function of SMN1 genes is associated with an expected lifespan of less than two years. In patients with type 2 or higher SMA phenotypes, symptom onset typically occurs later, often after important developmental milestones have been achieved. Among those with the less common type 3 SMA, symptoms might not develop until adolescence or even adulthood. This is largely related to the number of copies of the SMN2 gene, which produces less SMN protein than the SMN1 gene but produces enough to slow the onset and modify the severity of symptoms.
Neurologic Development Might Require Early Therapy
In SMA, particularly type 1, early diagnosis and early initiation of treatment is critical for permitting children to reach normal development milestones. In the studies with onasemnogene abeparvovec, type 1 SMA children have been initiated on treatment soon after symptoms or, in some cases, prior to symptoms. In an update on the LT-001 and LT-002 clinical research programs with onasemnogene abeparvovec, outcomes have been remarkable.
“[In patients up to 6.1 years of age,] we are seeing long-term persistence of gene function.”
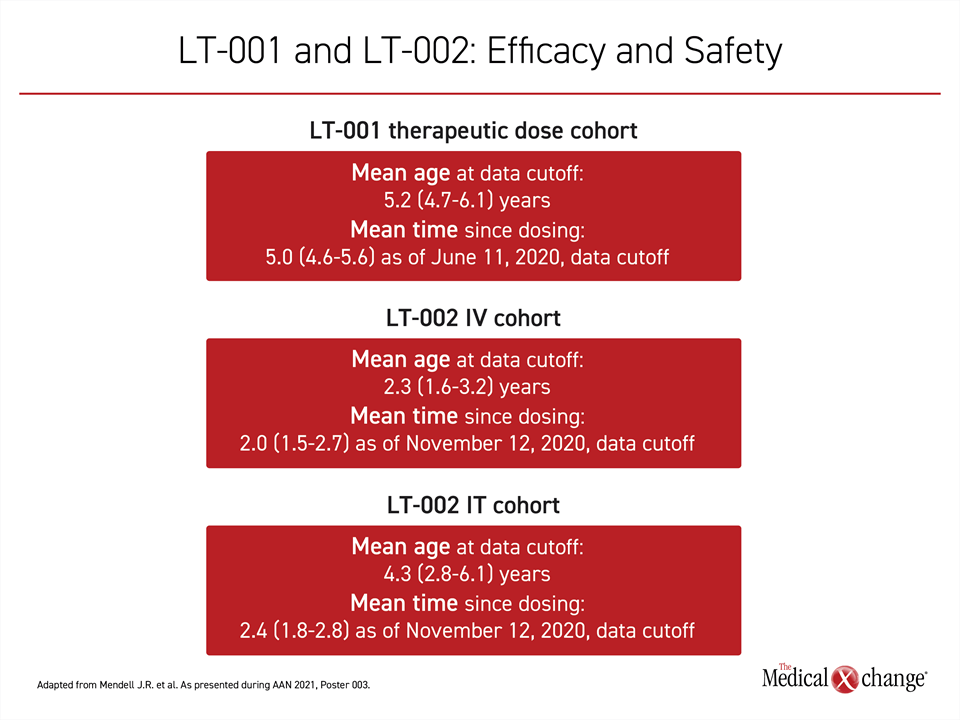
In the LT-001 program, which includes initial phase 1 data from the START trial, the mean age of the 12 patients who received a therapeutic dose is 5.2 years. The oldest is now 6.1 years, according to Dr. Jerry R. Mendell, Chair of Pediatric Research, Nationwide Children’s Hospital, Columbus, Ohio.
“We are seeing long-term persistence of gene function,” he said. For a group of children with an expected survival measured in months without treatment, “none of the children are on permanent ventilation.”
Long-Term Follow-up Reassuring
With the LT-002 program, there is now follow-up out to 2.7 years in a series of studies, including those that led to regulatory approval. Of the 22 symptomatic patients who participated in STR1VE, treatment was initiated on average at the age of 3.7 months. The majority of the patients have achieved development milestones within the window of time considered normal. In this follow-up, like the one of patients receiving therapeutic doses in LT-001, no patient has required long-term ventilatory support or other types of chronic life support, such as nasogastric feeding.
Safety data are also reassuring. According to Dr. Mendell, “There have been no emerging adverse safety signals in the long-term follow up for either the LT-001 or LT-002 programs.”
All of the studies in the LT-002 program are ongoing with planned follow-up for several additional years. In addition to STR1VE, which is testing gene therapy in symptomatic children, the SPR1NT study is evaluating initiation of treatment in presymptomatic patients. All of these studies have sufficient follow-up to provide strong evidence of efficacy and safety (Figure 4).
Due to the importance of maintaining adequate levels of the SMN protein, there is an advantage for a one-time dose gene replacement therapy over treatment that requires a lifetime of maintenance dosing. Although nusinersen, the first approved therapy for SMA, represented a vital breakthrough for control of SMA, newly reported real-world adherence data with this agent, which requires intrathecal injections every four months after completion of a loading-dose regimen, indicates that missed doses are common. This has implications for reaching and sustaining developmental milestones.
Adherence and Persistence Evaluated with Maintenance Therapy
In a U.S. prescribing database, SMA patients with type 1, 2, or 3 SMA who completed the loading dose phase with nusinersen were evaluated for adherence over an observation period in which at least four maintenance doses would have been administered under drug labelling. This included 23 patients with type 1 disease, 41 patients with type 2, and 260 patients with type 3.
“Patients who did not adhere were more likely to have SMA-related comorbidities across all SMA types, including feeding difficulties.”
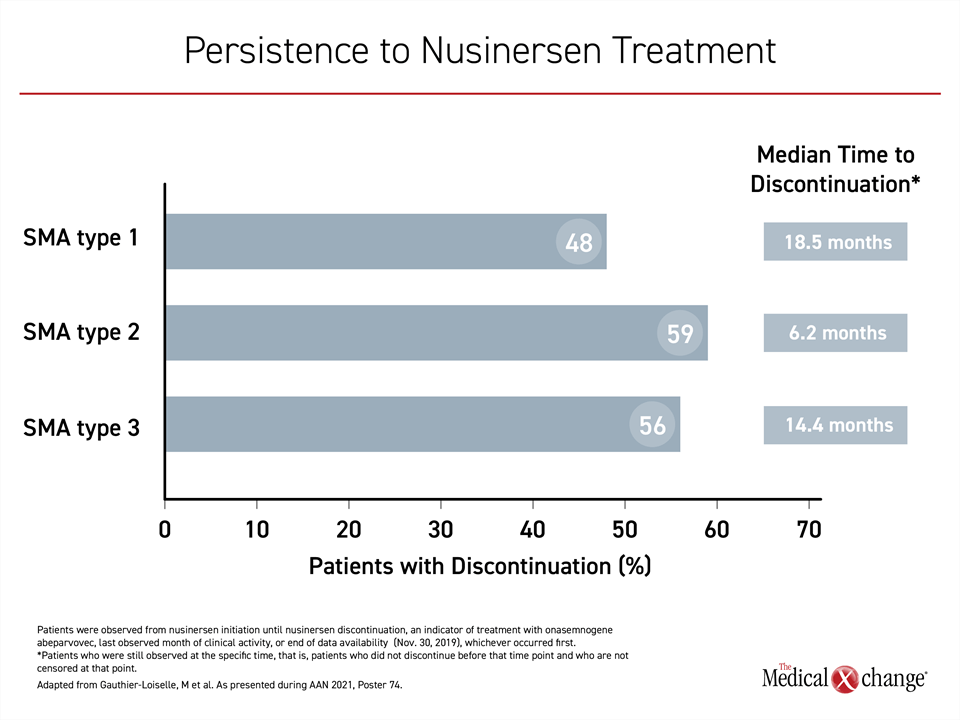
The proportion of patients receiving at least one dose off schedule, defined as >120 days between doses, was substantial, reaching 52.2%, 70.7%, and 53.1% for type 1, 2, and 3, respectively. The proportion with discontinuation over the course of follow-up was also significant, and this appeared to be relevant to outcomes (Figure 5).
“Patients who did not adhere were more likely to have SMA-related comorbidities across all SMA types, including feeding difficulties, dyspnea and other respiratory anomalies, failure to thrive, and muscle weakness,” reported Dr. Marjolaine Gauthier-Loiselle, Manager, Analysis Group, Montreal, Quebec.
The reasons for non-adherence to nusinersen could not be determined from data captured in real-world prescribing, but the investigators speculated that logistical challenges, such as travelling to site for multi-dose maintenance treatment, might have been among the causes. The discomfort of every-four-month intrathecal dosing is another. No cause-and-effect relationship can be determined for nonadherence and the greater risk of comorbidities in this retrospective analysis, but it is reasonable to consider that treatment delays and discontinuation “may result in loss of motor neuron function and disease progression,” Dr. Gauthier-Loiselle reported.
Gene Therapy with >2 SMN2 Copies
For SMA patients with only two copies of the SMN2 gene, gene therapy is particularly attractive because of the limits of stimulating adequate SMN protein production from therapies that target this gene. In the context of concerns about adherence, however, the efficacy of gene therapy in patients with more than two copies of SMN2 may also be relevant. In a summary of the 15 patients from the SPR1NT study with three copies of SMN2, event-free survival has been 100% after up to 21 months of follow-up.
“Thirteen of 15 children in the three-copy cohort were sitting independently. Almost all did so within the expected normal window of time for this milestone,” reported Dr. Kevin A. Strauss, Medical Director, Clinic for Special Children, Strasburg, Pennsylvania. The two children who did not yet reach this milestone have not yet exited the normal developmental window in which this activity is expected.
Eight of the 15 are standing, and six of the children have walked alone. None of the children that have yet to achieve these milestones have yet to exit, or in some cases enter, the normal developmental window for these activities.
“Thirteen of 15 children in the three-copy cohort were sitting independently. Almost all did so within the expected normal window of time for this milestone.”
“At their most recent visit, all 15 children in the three-copy cohort have gross motor performance on the Bayley-III test similar to healthy children in an age-matched healthy population,” Dr. Strauss said.
Conclusion
There are now three options for the treatment of SMA. The mechanisms of action differ. Unlike the maintenance agents nusinersen and the newly-approved risdiplam, onasemnogene abeparvovec requires a single infusion to restore SMN1 gene activity. These drugs have not been directly compared, but experienced reported in a survey that the time to improved motor function, as well as reversal of other deficits associated with SMA, occurred more rapidly if patients were initiated on onasemnogene abeparvovec than nusinersen. Motor function and development milestones were also achieved more quickly for those switched from nusinersen to onasemnogene abeparvovec than those maintained on nusinersen. Long-term, the single administration of onasemnogene abeparvovec circumvents issues of compliance. For children who participated in the first trials with onasemnogene abeparvovec, followup now exceeds five years with persistent benefit and favorable safety and tolerability.