neurologie
Conférence clinique et scientifique de 2022 de la Muscular Dystrophy Association (MDA)
Un lien est établi entre une thérapie génique opposée à l’amyotrophie spinale et un développement normal
Nashville – Les données définitives de l’étude SPR1NT, une étude de phase III menée chez des nourrissons atteints d’amyotrophie spinale (AS) de type 2 présentée lors du congrès annuel de la MDA, ont révélé qu’en corrigeant la production insuffisante d’une certaine protéine, une seule dose d’une thérapie génique administrée avant l’apparition des symptômes avait permis à ces nourrissons de franchir normalement les premières étapes de leur développement. Alors qu’il était à prévoir que sans traitement, la plupart d’entre eux ne réussissent pas à se tenir debout ni à marcher sans aide, ils y étaient tous parvenus au terme de 2 années de suivi. Des gains majeurs dans des cas d’AS de type 1, la forme la plus grave de la maladie, ont aussi été rapportés lors du congrès.
Encore récemment, il n’existait pas de traitement efficace pour empêcher l’évolution de l’AS, une maladie souvent mortelle pendant la petite enfance. Le premier agent dirigé contre cette maladie, le nusinersen, un oligonucléotide antisens qui doit être injecté tous les 4 mois par voie intrathécale dans le cadre d’un traitement d’entretien, a été homologué il y a près de 5 ans. L’onasemnogène abéparvovec, seul agent de thérapie génique à l’heure actuelle, a été homologué à la fin de 2021 au Canada et a été suivi peu de temps après par le risdiplam, un modificateur d’épissage du gène SMN2. Prise tous les jours par voie orale, cette petite molécule, tout comme le nusinersen, stimule la production de protéines SMN dont la production est insuffisante dans les cas d’AS, mais elle exige aussi un traitement d’entretien.
Pour une entrevue exclusive avec le Dr Alex MacKenzie couvrant l’impact sur la pratique clinique, cliquez ici
La thérapie génique dans le traitement de l’AS chez des enfants porteurs de 3 copies du gène SMN2
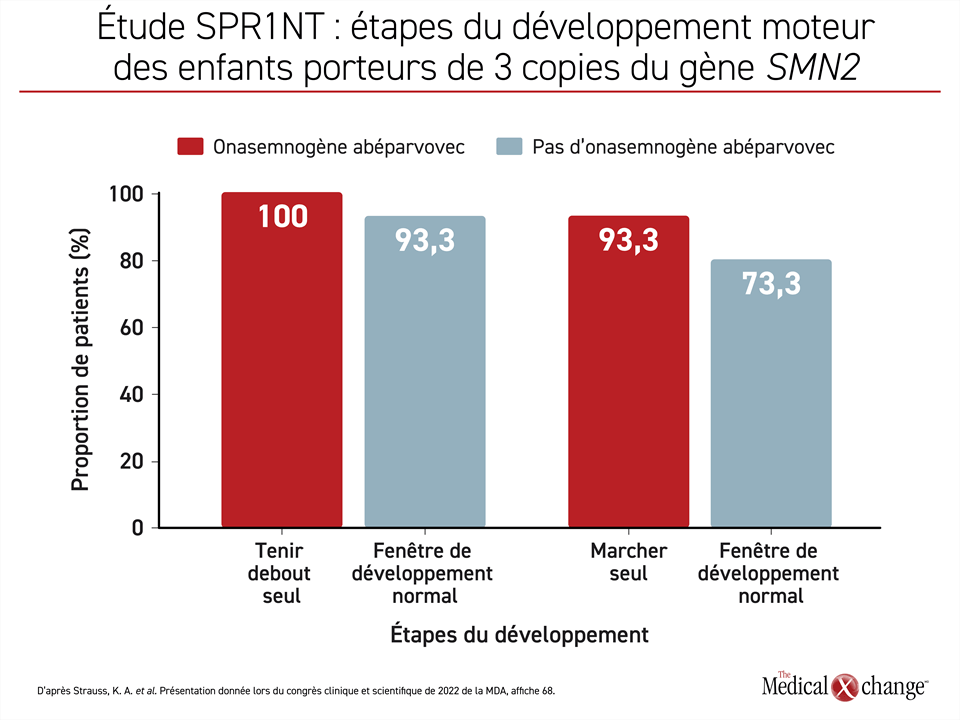
Le Dr Kevin Strauss, de la Clinic for Special Children, de Strasburg, en Pennsylvanie, et chercheur principal de l’étude de phase III SPR1NT, a affirmé que cette étude visait à évaluer l’efficacité et l’innocuité d’une seule dose d’onasemnogène abéparvovec injectée par voie intraveineuse pendant la phase présymptomatique de l’AS à 15 nourrissons porteurs de 3 copies du gène SMN2. Le paramètre d’évaluation principal était la capacité de se tenir debout sans aide, mais beaucoup d’autres étapes du développement ont été surveillées pendant la période de suivi. En substance, les chercheurs voulaient savoir si l’établissement rapide d’un diagnostic et l’amorce d’un traitement pendant la phase présymptomatique de la maladie permettaient d’empêcher l’apparition des symptômes et pas seulement de la retarder dans ce sous-groupe de patients.
« Les 15 enfants (100 %) sont toujours en vie et peuvent se tenir debout seuls », a déclaré le Dr Strauss. Quatorze d’entre eux y étaient parvenus en 16,9 mois, soit à un âge qui se situe à l’intérieur des valeurs de référence établies par l’Organisation mondiale de la santé (OMS).
Quatorze des enfants ont également réussi à marcher sans aide avant la fin de la période de suivi, 11 d’entre eux y étant parvenus à un âge normal selon l’OMS (Figure 1). De plus, 11 des enfants étaient capables de s’asseoir sans aide à un âge normal, une étape qui survient plus tôt dans le développement. Aucun des enfants n’a eu besoin de quelque forme que ce soit de ventilation assistée ni d’être nourri autrement que par la bouche pendant cette étude.
Innocuité et absence d’effets indésirables graves démontrées
Aucun effet indésirable grave imputable au traitement ne s’est produit. Une hausse des concentrations d’enzymes hépatiques et une thrombopénie observées respectivement chez 4 et 2 patients comptaient parmi les effets indésirables sans gravité. La hausse des résultats aux épreuves fonctionnelles hépatiques était de grade 3 chez un seul des enfants ayant obtenu des résultats anormalement élevés, mais aucun d’eux n’a subi de conséquence sérieuse comme un ictère ou une encéphalopathie hépatique. Les 2 cas de thrombopénie, aussi décelés dans le cadre de la surveillance biochimique, n’ont pas eu non plus de conséquences cliniques ni induit des changements généraux durant le suivi qui a comporté notamment des échocardiogrammes et autres moyens de surveillance de la fonction cardiaque.
Dans l’ensemble, le temps écoulé avant le diagnostic était de 9,9 jours dans ce groupe de nourrissons dont la maladie a été découverte le plus souvent grâce à un test de dépistage à la naissance. Tous ces enfants sont nés après la 37e semaine de gestation, sauf un. Ils avaient 28,7 jours en moyenne quand ils ont reçu la thérapie génique; le plus âgé avait 43 jours. Aucun n’avait encore montré des symptômes de l’AS.
« Tous les participants à l’étude ont marqué des gains constants sur l’échelle III de Bayley pour la motricité. »
Faisant référence à un outil normalisé servant à suivre le développement des enfants, le Dr Strauss a affirmé : « Tous les participants à l’étude ont marqué des gains constants sur l’échelle III de Bayley pour la motricité ». Il a ajouté qu’après comparaison avec des enfants indemnes d’AS appariés en fonction de l’âge, « les scores bruts obtenus par les participants sur l’échelle III de Bayley pour la motricité fine et globale se situaient à l’intérieur des valeurs normales ».
En administrant le traitement tôt, il pourrait être possible d’empêcher la maladie d’évoluer
Cette étude étaye l’hypothèse voulant qu’en administrant la thérapie génique tôt, avant même que les symptômes n’apparaissent, il soit possible aux enfants atteints d’AS, une maladie imputable à une mutation du gène SMN1, de franchir les premières étapes du développement moteur.
C’est un point important parce que l’onasemnogène abéparvovec remplace le gène SMN1 muté qui code pour la protéine SMN, une protéine essentielle à la survie du motoneurone. La perte de fonction du gène SMN1 est la cause sous-jacente de l’AS. Un deuxième gène, le gène SMN2, peut aussi coder pour la protéine SMN, mais elle est produite en quantités nettement inférieures. Même si le nusinersen et le risdiplam amplifient la production de la protéine SMN par la voie du gène SMN2, ils ne permettent pas de traiter le processus pathologique sous-jacent.
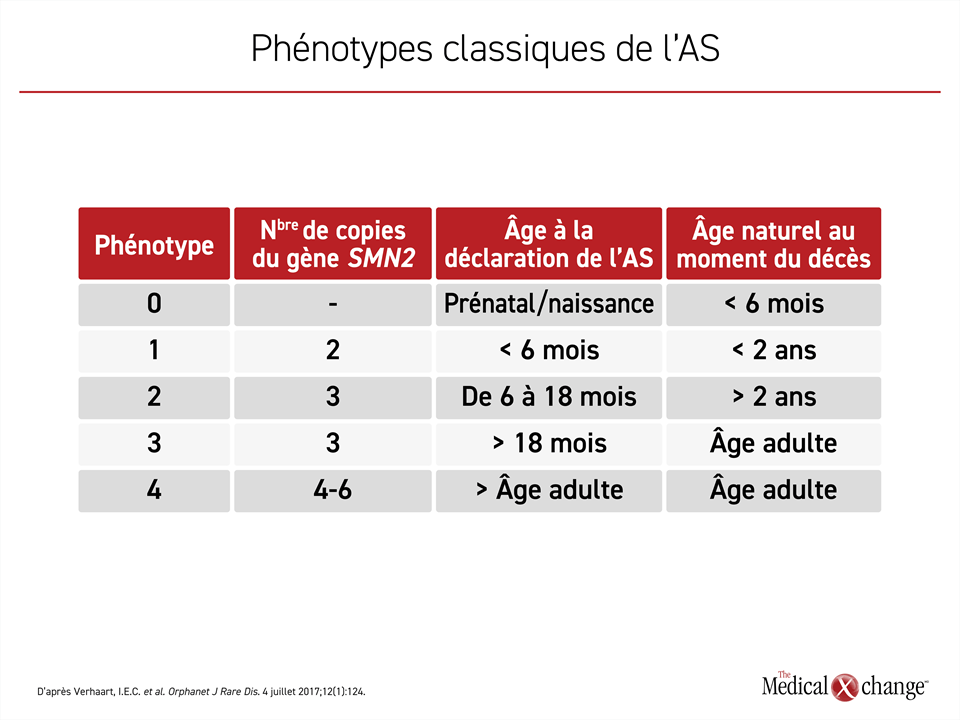
Les patients recrutés dans l’étude SPR1NT étaient porteurs de 3 copies du gène SMN2. Le nombre de copies de ce gène détermine la gravité de la maladie. Les enfants atteints d’AS de type 1, la forme la plus répandue et la plus grave de cette maladie, sont porteurs de 2 copies seulement du gène SMN2. Bien que la maladie évolue plus lentement chez les enfants porteurs de 3 copies du gène SMN2, le pronostic reste sombre, car peu d’entre eux survivent à la petite enfance. Chez les porteurs de 4 copies du gène SMN2 ou plus, qui sont rares, il se peut que la présence de l’AS ne soit pas décelée avant l’âge adulte (Tableau 1).
Traitement de fond de l’AS de type 1 et répercussions sur les fonctions bulbaires
Des études sont actuellement menées sur la thérapie génique dirigée contre l’AS de type 1, notamment deux études de phase III, intitulées STR1VE-US et STR1VE-EU, au cours desquelles l’onasemnogène abéparvovec est administré pendant la petite enfance. Une analyse a posteriori des données combinées de ces deux études et d’une étude de phase I appelée START, qui a été présentée devant la MDA, a servi à examiner plus précisément l’effet de la thérapie génique sur la préservation des fonctions bulbaires, dont certaines comptent parmi les fonctions vitales les plus élémentaires.
Les fonctions du motoneurone de la région bulbaire doivent absolument être préservées parce qu’elles « régissent les muscles permettant d’ouvrir la bouche, de mastiquer, d’avaler et de parler », a expliqué la principale responsable de cette analyse, la Dre Katlyn McGrattan, de l’Université du Minnesota, à Minneapolis. La détérioration de ces neurones est associée à l’étouffement, aux infections et à la mort.
« Les fonctions du motoneurone de la région bulbaire doivent absolument être préservées parce qu’elles régissent les muscles permettant d’ouvrir la bouche, de mastiquer, d’avaler et de parler. »
L’analyse a posteriori a porté sur les données de 54 participants aux deux études de phase III et de 11 participants à l’étude START. Les résultats obtenus pour les paramètres évalués n’étaient pas connus pour tous les patients de toutes les études, mais les chercheurs voulaient compiler les données déjà disponibles. Les fonctions bulbaires analysées à partir des examens cliniques et de tests normalisés, tels que les échelles de Bayley, ont été les suivantes : capacité à communiquer avec un inconnu, à avaler des aliments et des liquides, et à protéger leurs voies respiratoires.
La plupart des fonctions bulbaires étaient opérantes
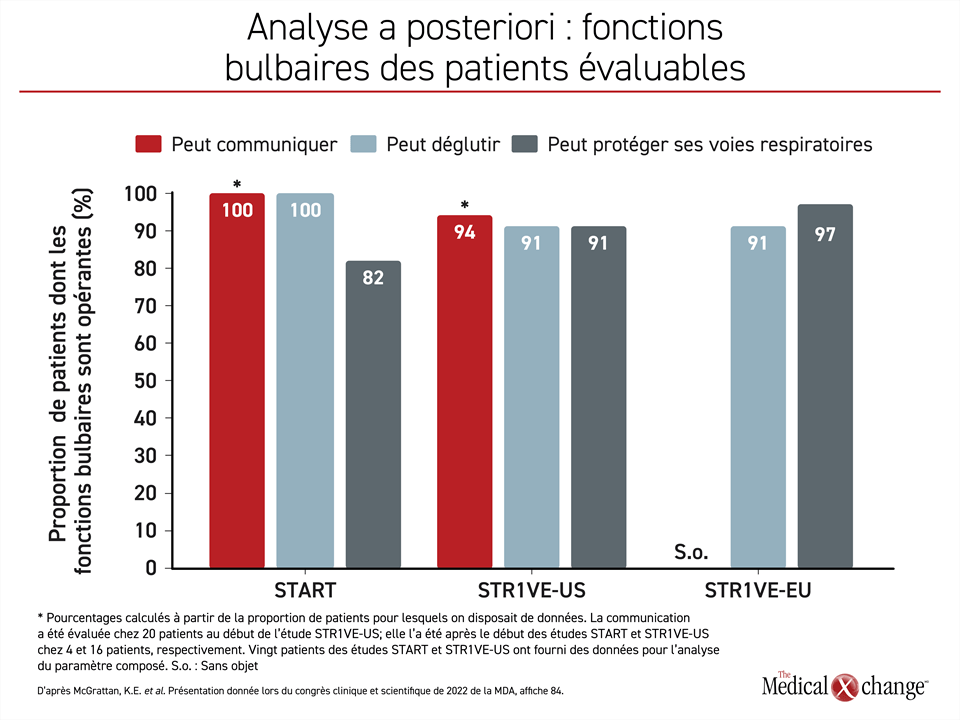
Même si certaines de ces fonctions étaient initialement déficitaires, elles étaient toutes opérantes chez la plupart des patients évaluables à un moment ou à un autre après qu’ils aient reçu la thérapie génique. L’aptitude à communiquer n’est pas évaluée pendant l’étude STR1VE-EU, mais 14 (94 %) des 15 sujets de l’étude STR1VE‑US et les 4 sujets (100 %) de l’étude START évalués en sont devenus capables.
Les 11 patients (100 %) de l’étude START évalués en sont venus à déglutir normalement, tout comme 20 (91 %) des 22 patients de l’étude STR1VE-US et 29 (91 %) des 32 patients de l’étude STR1VE-EU évalués. Neuf (82 %) des 11 patients de l’étude START, 20 (91 %) des 22 patients de l’étude STR1VE-US et 31 (97 %) des 32 patients de l’étude STR1VE-EU évalués étaient capables de protéger leurs voies respiratoires (Figure 2).
Il ressort de l’analyse de ces données combinées que chacune de ces trois étapes du développement a été franchie par plus de 90 % des patients évaluables. L’analyse de ces trois paramètres réunis révèle que 16 (80 %) des 20 patients évaluables s’en sont montrés capables avant la fin de la période de suivi de chacune des études, qui a duré 24 mois pour l’étude START et 18 mois pour les études STR1VE-US et STR1VE-EU. La plupart des enfants atteints d’AS de type 1 ayant participé à ces études n’ont pas eu besoin d’alimentation assistée pendant ces études.
À la lumière de cette analyse a posteriori, la thérapie génique semble efficace pour soutenir les fonctions élémentaires chez les enfants atteints d’AS de type 1 si elle est administrée tôt. Selon la Dre McGrattan, les données indiquent qu’elle pourrait transformer « une démarche palliative en une démarche réadaptative ».
Les patients atteints d’AS de type 1 ont atteint les objectifs principaux
Une fois combinées, ces données « indiquent que les patients atteints d’AS de type 1 symptomatique traités avec de l’onasemnogène abéparvovec pouvaient communiquer suffisamment, déglutir, satisfaire leurs besoins nutritionnels et protéger leurs voies respiratoires », a-t-elle précisé. L’effet de la thérapie génique sur les fonctions bulbaires n’avait pas encore été évalué de façon approfondie, mais ces données établissent un parallèle entre l’administration d’une seule dose d’onasemnogène abéparvovec dès le début de l’AS de type 1 et une stabilisation des fonctions motrices qui sont souvent reliées à un décès prématuré.
« Les patients atteints d’AS de type 1 symptomatique [traités]… pouvaient communiquer suffisamment, déglutir et satisfaire leurs besoins nutritionnels. »
Analyse menée en pratique clinique sur un éventail plus large de patients aux prises avec l’AS
Une analyse réalisée en pratique clinique dans le cadre d’une enquête communautaire menée en 2020 par Cure SMA confirme l’utilité de l’onasemnogène abéparvovec, même s’il est administré après l’âge de 6 mois. Lors de cette étude de cohorte non interventionnelle, qui a aussi fait l’objet d’une présentation durant le congrès annuel de 2022 de la MDA, 53 nourrissons atteints d’AS ayant reçu une dose d’onasemnogène abéparvovec entre l’âge de 6 et 23 mois ont été comparés à 53 autres enfants appariés en fonction de leur âge, mais qui n’en avaient pas reçu.
La plupart des enfants des deux groupes étaient atteints d’AS de type 1, les autres étant surtout aux prises avec le type 2 de la maladie. Peu de patients étaient atteints du type 3 ou d’un type d’AS qui n’avait pas été précisé.
Franchissement plus marqué des étapes du développement et utilisation moindre des ressources de santé
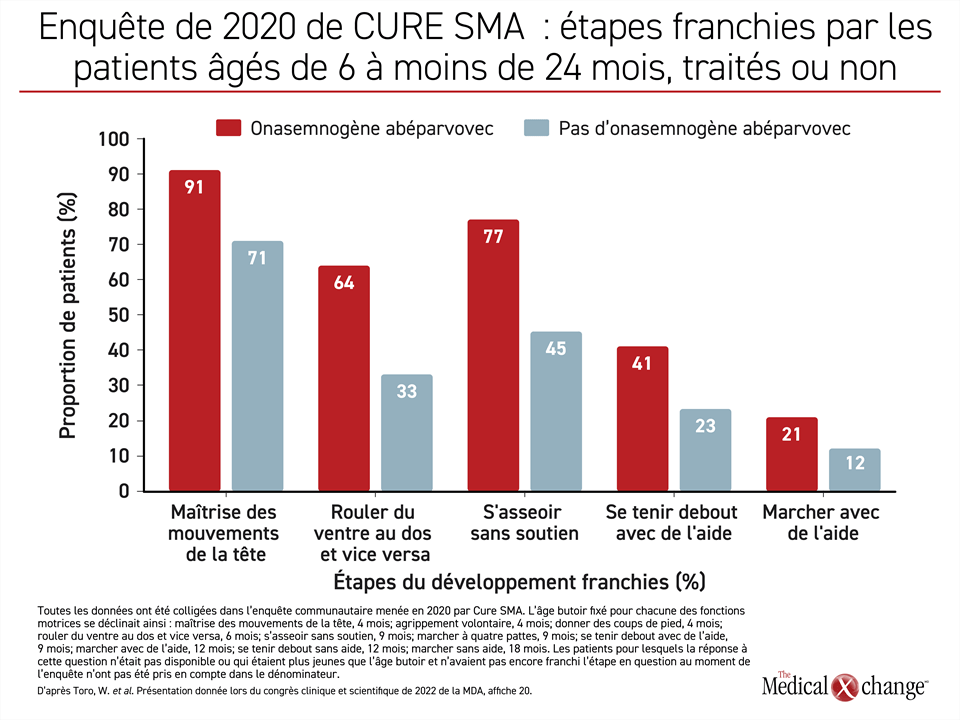
Les résultats de l’enquête de CURE SMA ont plaidé en faveur de l’onasemnogène abéparvovec pour pratiquement tous les paramètres évalués, soit le franchissement des étapes du développement, l’absence de complications de l’AS et la qualité de vie. Cet organisme de défense des droits des patients gère une base de données autodéclarées depuis 2017. Les personnes interrogées étaient des proches aidants de patients atteints d’AS. Analysis Group, de Boston, au Massachusetts, a été chargé de l’organisation de cette enquête.
Selon les réponses qui y ont été colligées, « les patients traités par l’onasemnogène abéparvovec ont été plus nombreux [que ceux qui n’en ont pas reçu] à franchir chacune des étapes évaluées : maîtriser les mouvements de la tête, rouler du ventre au dos et vice versa, s’asseoir sans soutien, se tenir debout avec de l’aide, marcher avec de l’aide, se tenir debout sans aide et marcher sans aide », a affirmé la Dre Min Yang, experte en économie de la santé et vice-présidente d’Analysis Group (Figure 3).
De plus, les enfants traités par l’onasemnogène abéparvovec avaient été hospitalisés moins souvent au cours des 12 mois précédents que ceux qui n’avaient pas reçu cet agent (32 % vs 36 %) et ils risquaient moins d’avoir besoin d’une trachéotomie et de ventilation assistée (6 % vs 15 %), et d’avoir besoin de ventilation assistée pendant plus de 16 heures par jour (6 % vs 30 %), respectivement. La Dre Yang a ajouté que les enfants traités par la thérapie génique avaient utilisé moins de ressources de santé.
Se fondant sur les résultats enregistrés sur le questionnaire EQ-5D sur la qualité de vie, la Dre Yang a déclaré qu’en général, les nourrissons traités par la thérapie génique ont connu un sort plus favorable que ceux qui ne l’avaient pas été; ils ont notamment éprouvé moins de douleurs et d’anxiété, et ils ont plus été en mesure de s’adonner aux activités de la vie quotidienne. Elle a ajouté que ces avantages relatifs étaient généralement constants, indépendamment du nombre de copies du gène SMN2. Les deux patients porteurs de 4 copies de ce gène traités après l’âge de 6 mois ont franchi toutes les étapes du développement propres à leur âge sans que des symptômes graves motivent leur hospitalisation.
Conclusion
La thérapie génique à base d’onasemnogène abéparvovec, qui s’administre en une seule dose, est un moyen très efficace de marquer des gains soutenus chez les enfants atteints d’AS. Si elle est administrée pendant la phase présymptomatique des formes graves de la maladie, elle permet à la majorité des enfants de franchir les diverses étapes de leur développement à un âge normal. Lors de l’étude de phase III SPR1NT récemment terminée et dans laquelle des porteurs de 3 copies du gène SMN2 ont été recrutés, 14 des 15 participants sont maintenant capables de marcher de façon autonome. Une analyse a posteriori des données colligées chez des enfants atteints d’AS de type 1, la forme la plus grave de la maladie, a confirmé la concrétisation des fonctions fondamentales de la vie, telles que la déglutition et la communication. La thérapie génique pourrait bien transfigurer le pronostic de l’AS.