Neurology
2022 Muscular Dystrophy Association (MDA) Clinical & Scientific Conference
Gene Replacement Therapy for Spinal Muscular Atrophy Associated with Normal Development
Nashville – By restoring production of a deficient protein, a single pre-symptomatic dose of a gene replacement therapy permits development of normal early milestones in infants with spinal muscular atrophy (SMA), according to final data from a phase III trial presented at the MDA annual meeting. The study, called SPR1NT, enrolled infants with SMA type 2. Most of these children would not have been expected to stand or walk independently without therapy, but at 2 years of follow-up all children reached this milestone. Major benefits from gene replacement therapy in SMA type 1, the most severe form of this disorder, were also reported here.
Until recently, there were no effective treatments to prevent progression of SMA, which is commonly fatal in early childhood. The first therapy, nusinersen, an antisense oligonucleotide that requires maintenance intrathecal doses every 4 months, was approved nearly 5 years ago. Onasemnogene abeparvovec, the only current gene replacement therapy, was approved in Canada at the end of 2021. Risdiplam, an oral small-molecule SMN2 splicing modifier was approved soon after. Taken daily, risdiplam, like nusinersen, increases availability of the deficient SMN protein in SMA but also requires maintenance dosing.
For an exclusive interview with Dr. Alex MacKenzie on the impact to clinical practice, click here
Gene Replacement Therapy for SMA with Three SMN2 Gene Copies
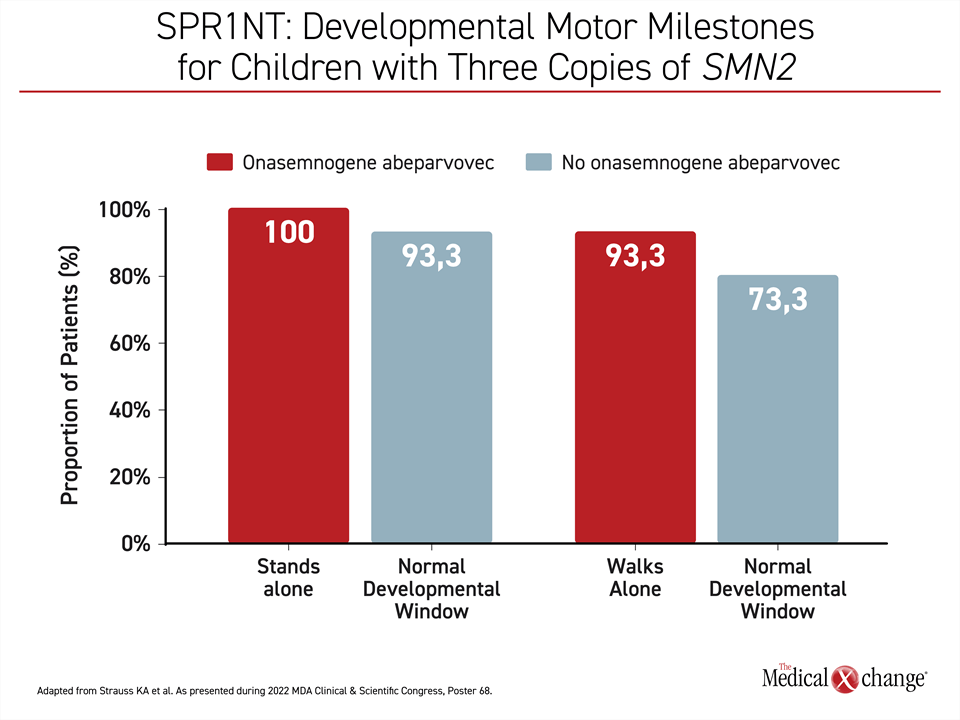
The objective of the phase 3 SPR1NT trial was to evaluate the safety and efficacy of onasemnogene abeparvovec in SMA infants with three copies of the SMN2 gene, according to lead investigator, Dr. Kevin Strauss, Clinic for Special Children, Strasburg, Pennsylvania. The single intravenous dose was administered in 15 infants prior to the onset of symptoms. Ability to stand independently was the primary endpoint but numerous other milestones were monitored over the course of follow-up. Ultimately, the goal is to determine whether early diagnosis and pre-symptomatic treatment of this SMA subgroup can prevent symptom onset, not just delay it. Safety was also assessed.
“All 15 children—100%—are alive and all can stand independently,” reported Dr. Strauss. Fourteen of the 15 achieved this milestone within 16.9 months, which is within the normal developmental window as defined by the World Health Organization (WHO).
Fourteen were also able to walk independently by the end of follow-up, 11 of which reached this milestone within the WHO-defined normal period (Figure 1). Earlier milestones were also reached with 11 of the children sitting independently at the appropriate age. No child in this study required any form of ventilatory support or any form of non-oral feeding, at any time.
Safety Demonstrated with No Serious Adverse Events
There were no serious treatment-related adverse events. Non-serious events included liver enzyme elevations in 4 patients and thrombocytopenia in 2 patients. Of those with elevated liver function tests, only one was grade 3, but there were no serious consequences, such as jaundice or hepatic encephalopathy, in any patient. The 2 cases of thrombocytopenia, also detected on laboratory monitoring, did not lead to clinical consequences or systemic changes on follow-up that included echocardiograms and other cardiac function monitoring.
“All study participants developed steady gains in the Bayley-III motor scales.”
Overall, the mean time to diagnosis was 9.9 days in a group that was largely diagnosed through newborn screening. The gestational age was greater than 37 weeks in all children but one. The mean age at the time of gene replacement therapy was 28.7 days. The oldest child at treatment start was 43 days. None had yet developed symptoms of SMA.
“All study participants developed steady gains in the Bayley-III motor scales,” said Dr. Strauss, referring to a standardized tool to monitor childhood development. When compared to non-affected age-matched peers, “the raw Bayley-III fine and gross motor scores fell within the normal range,” he added.
Early Treatment May Prevent Progression
The study supports the hypothesis that early, pre-symptomatic delivery of gene replacement therapy can prevent loss of early motor milestones in SMA, a disease resulting from a mutation in the SMN1 gene.
This is important because onasemnogene abeparvovec replaces the mutated SMN1 gene encoding the SMN1 protein responsible for motor neuron survival. Loss of SMN1 function is the underlying defect of SMA. A second gene, SMN2, can also encode SMN1 protein, but it does so in far reduced quantities. While both nusinersen and risdiplam increase production of the SMN1 protein through the SMN2 gene pathway, they do not treat the underlying pathology.
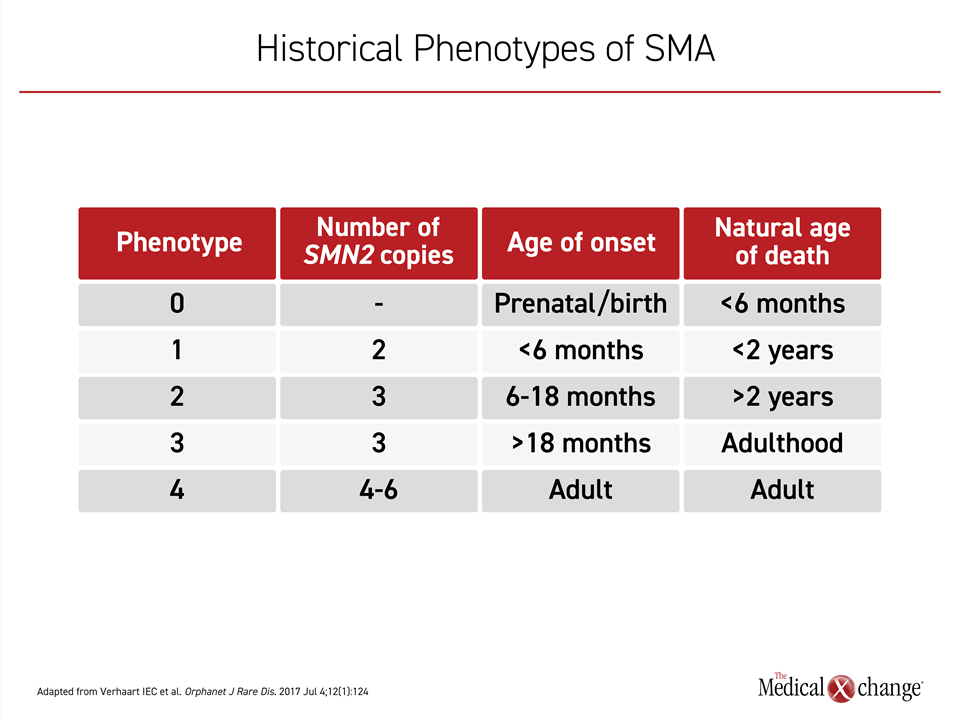
In the SPR1NT study, patients were enrolled with 3 SMN2 gene copies. The number of SMN2 gene copies determines severity. Children with SMA type 1, the most common and severe form, have just two SMN2 gene copies. Even though the progression in patients with three SMN2 copies is generally slower, the prognosis remains poor; few children survive early childhood. In SMA defined by 4 or more SMN2 gene copies, which is uncommon, the presence of SMA might not be detected until adulthood (Table 1).
Disease-Modifying Therapy in SMA Type 1 and Impact on Bulbar Function
Studies of gene therapy in SMA type 1 are ongoing. These include two phase 3 trials in which onasemnogene abeparvovec is being initiated early in childhood. In a post-hoc analysis presented at the MDA, data from these two trials, called STR1VE-US and STR1VE-EU, plus a phase 1 trial called START were combined to specifically explore the impact of gene replacement therapy on preserving bulbar function, which involves some of the most basic functions of life.
Preservation of bulbar motor neuron function is critical because they “control muscles required for mouth opening, chewing, swallowing and speaking,” explained lead investigator of this analysis, Dr. Katlyn McGrattan, University of Minnesota, Minneapolis. The deterioration of these neurons is associated with choking, infection, and death.
“Preservation of bulbar neuron function is critical because they control muscles required for mouth opening, chewing, swallowing and speaking.”
The post-hoc analysis included data on 54 patients from the two phase III trials and 11 patients from START. Endpoint assessments were not available on all patients in all studies, but the goal was to collate those data that were available. Using clinical examination as well as standardized tests, such as the Bayley scales, bulbar function was defined as the ability to communicate with an unknown listener, ability to swallow food and liquids, and ability to maintain airway protection.
Most Bulbar Functions Achieved
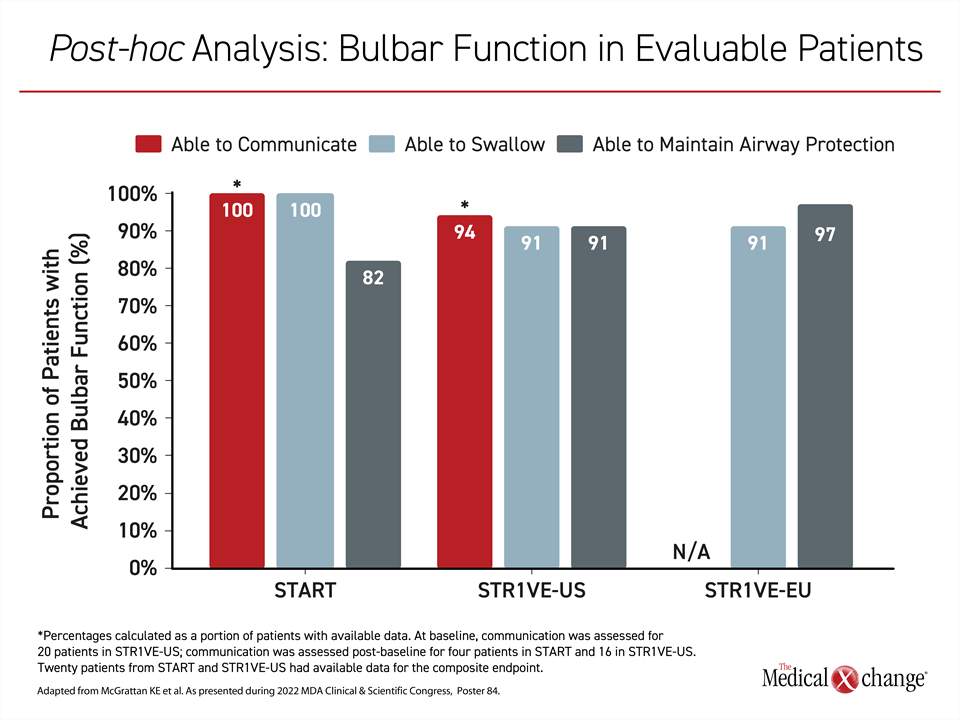
Despite deficits in some of these functions at baseline, most evaluable patients achieved each of these bulbar functions at some point after receiving gene replacement therapy. Ability to communicate was not assessed in STR1VE-EU, but it was achieved at some point in 14 (94%) of the 15 patients evaluated in STR1VE-US and all 4 (100%) of the patients evaluated in START.
Normal swallowing was achieved at some point in 11 (100%) of the 11 patients evaluated in the START, 20 (91%) of the 22 patients evaluated in STR1VE-US, and 29 (91%) of the 32 patients evaluated in STR1VE-EU. Ability to maintain airway protection was achieved at some point in 9 (82%) of the 11 patients evaluated in START, 20 (91%) of the 22 patients evaluated in STR1VE-US and 31 (97%) of the 32 patients evaluated in STR1VE-EU (Figure 2).
In a combined analysis, each of these three milestones was achieved in more than 90% of patients who were evaluable. When assessed as a composite of all three endpoints, 16 (80%) of the 20 evaluable patients reached all these measures at some point in the study by the end of follow-up, which was 24 months in START and 18 months in STR1VE-US and STR1VE-EU. Most of the SMA type 1 children in these studies did not require feeding support at any time during the study.
Based on this post-hoc analysis, gene replacement therapy appears to be effective in sustaining basic function in SMA type 1 children if initiated early. According to Dr. McGrattan, the data indicate that this type of therapy can move treatment of SMA “from a palliative approach to a rehabilitative approach.”
Key Outcomes Reached in SMA Type 1 Patients
The combined data “indicate that patients with symptomatic SMA type 1 treated with onasemnogene abeparvovec could communicate sufficiently, swallow and meet nutritional needs, and maintain airway protection,” she said. Previously, the effect of gene replacement therapy on bulbar function has not been comprehensively evaluated, but these data associate a one-time dose of onasemnogene abeparvovec early in the course of SMA type 1 with a stabilization of motor functions that are often associated with early death.
“[Treated] patients with symptomatic SMA type 1… could communicate sufficiently, swallow and meet nutritional needs.”
Real-world Analysis on Broader Range of SMA Patients
A real-world analysis drawn from the 2020 Cure SMA Community Update Survey supports the value of onasemnogene abeparvovec even when initiated after 6 months of age. In this non-interventional cohort study also presented at the 2022 MDA annual meeting, 53 infants with SMA that received a dose of onasemnogene abeparvovec between the ages of 6 and 23 months were compared to 53 infants with SMA matched for age who did not receive onasemnogene abeparvovec.
The majority of patients in both groups had SMA type 1. Most of the remaining patients had SMA type 2. A small percentage of patients had SMA type 3 or the type of SMA was unspecified.
Greater Achievement of Motor Milestones and Lower Healthcare Resource Utilization
For essentially every outcome evaluated, which included achievement of developmental milestones, freedom from SMA complications, and quality of life, onasemnogene abeparvovec was favored, according to results of the survey, which was conducted by CURE SMA. This patient advocacy organization has maintained a self-reported database since 2017. Those completing the survey were caregivers of SMA patients. The study was organized by the Analysis Group, Boston, Massachusetts.
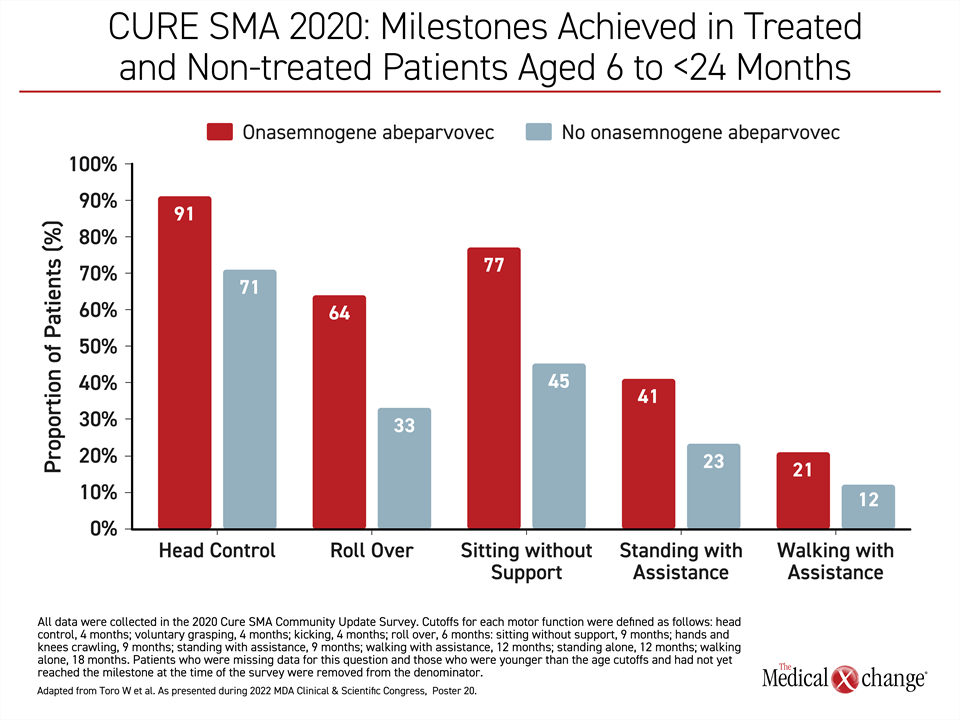
Based on the survey responses, “more onasemnogene abeparvovec-treated patients [than those not on gene replacement therapy] achieved each measured milestone, including head control, rolling over, sitting without support, standing with assistance, walking with assistance, standing alone, and walking alone,” reported Dr. Min Yang, health economics expert and Vice-president, Analysis Group, Boston, Massachusetts (Figure 3).
In addition, patients treated with onasemnogene abeparvovec had fewer hospitalizations in the prior 12 months compared with the non–onasemnogene abeparvovec-treated group (32% vs. 36%), were less likely to require tracheostomy with ventilator (6% vs. 15%) and were less likely to require respiratory support for > 16 hours per day (6% vs. 30%), respectively. Dr. Yang also reported that those receiving gene replacement therapy required less healthcare-related resources.
On the EQ-5D tool for evaluating quality of life, infants receiving gene replacement therapy had generally better outcomes, such as less pain, less anxiety, and greater ability to perform activities of daily living than those who were not treated with onasemnogene abeparvovec, according to Dr. Yang. She reported that these relative advantages were generally consistent across SMN2 gene copy number. Both patients with four SMN2 gene copies treated after 6 months of age achieved all the age-appropriate milestones without serious symptoms leading to hospitalization.
Conclusion
The gene replacement therapy onasemnogene abeparvovec, which is administered in a single dose, is highly effective for providing sustained benefits in children with SMA. If initiated pre-symptomatically in severe forms of this disease, developmental milestones are achieved in most children within the age-appropriate timeframe. In the newly completed phase 3 SPR1NT trial, which enrolled patients with 3 SMN2 gene copies, 14 of the 15 infants enrolled are walking independently. Key functions of life, such as swallowing and communicating, were observed in a post-hoc analysis of patients with SMA type 1, the most severe form. The gene replacement therapy appears to be transforming the prognosis of SMA.