neurologie
Congrès annuel de 2022 sur l’AS ꟷ recherche et soins cliniques
Le rapport intitulé State of SMA confirme les résultats nettement meilleurs obtenus dans le traitement de l’amyotrophie spinale
Anaheim – Une analyse de données contemporaines a révélé que la mortalité des patients atteints d’amyotrophie spinale (AS) a beaucoup baissé depuis l’arrivée de nouveaux traitements il y a 5 ans. Publiée dans State of SMA, un document remis aux participants au congrès annuel de 2022 sur la recherche sur l’AS et les soins cliniques prodigués contre cette maladie, cette analyse nous apprend que l’amélioration des résultats obtenus dans le traitement de l’AS, dont la survie, s’étend maintenant à des formes graves au point d’être inéluctablement mortelles. Il y est également question d’analyses a posteriori de l’ensemble des données tirées de trois études sur la préservation et la stabilisation du fonctionnement bulbaire, jumelées aux données définitives de l’étude SPR1NT, qui décrivaient les gains majeurs observés avec la thérapie génique chez des nourrissons présymptomatiques.
Si les trois agents actuellement offerts contre l’AS sont efficaces, c’est parce qu’ils peuvent contrer la cause de cette maladie, un déficit en protéine de survie du motoneurone (SMN). Deux de ces agents exigent un traitement d’entretien, alors que l’agent de thérapie génique rétablit la production de cette protéine. L’homologation du premier de ces agents, qui doit être administré de façon répétée par voie intrathécale, remonte à 5 ans. L’agent de thérapie génique à dose unique et l’agent à prise orale quotidienne ont été homologués par la suite.
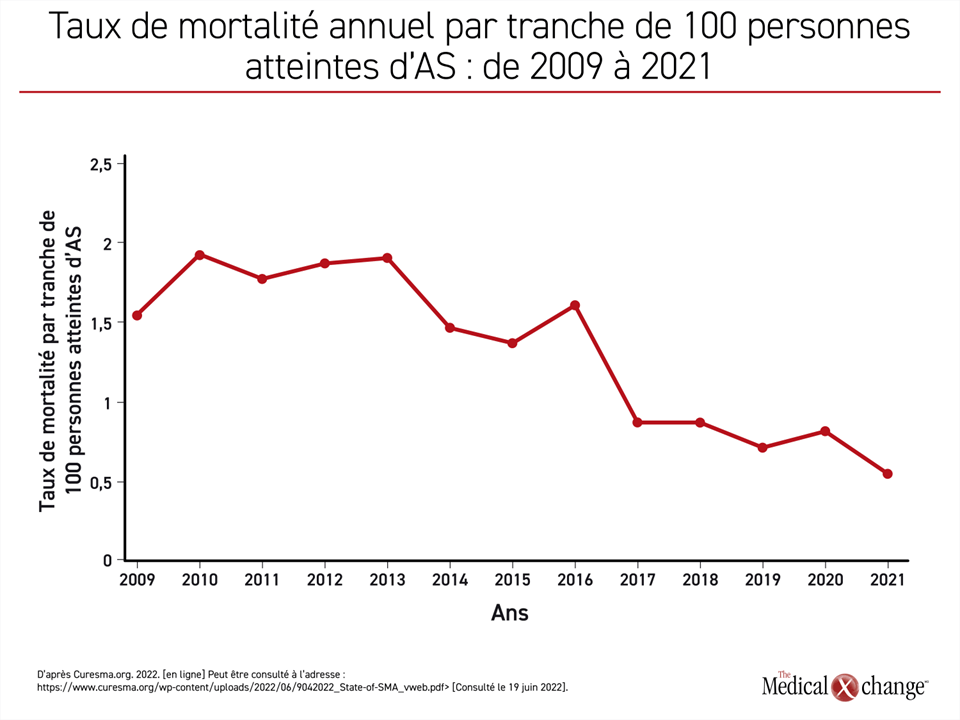
Depuis l’arrivée de ces traitements, le nombre de décès par tranche de 100 patients atteints d’AS a chuté de près de 75 %, passant d’un taux annuel de presque 2,0/100 patients il y a dix ans à 0,5 en 2021 (Figure 1). Les améliorations observées à d’autres niveaux, tel que l’invalidité, ont été du même ordre, selon les auteurs du rapport, dont la Dre Mary Schroth, directrice médicale de Cure SMA, et une équipe d’analystes de données.
De nouvelles réductions de la mortalité causée par l’AS sont à prévoir
L’amélioration des résultats pourrait bien se poursuivre. Comme le taux de dépistage augmente rapidement chez les nouveau-nés, on pourra intervenir plus tôt dans la vie. « Grâce à l’augmentation du dépistage chez les nouveau-nés, il arrive moins souvent que le diagnostic soit posé après l’apparition des symptômes », ont expliqué la Dre Schroth et ses collègues auteurs. En dépistant et en traitant la maladie avant l’apparition des symptômes, il est possible d’éviter les déficits neurofonctionnels irréversibles.
« Grâce à l’augmentation du dépistage chez les nouveau-nés, il arrive moins souvent que le diagnostic soit posé après l’apparition des symptômes. »
Tous les traitements accroissent la quantité de protéine SMA. Si les patients atteints d’une forme légère d’AS sont la minorité, il demeure que la plupart d’entre eux sont aux prises avec une maladie grave caractérisée par un petit nombre de copies du gène SMN2, qui les emporte en moins de 2 ans s’ils ne sont pas traités. Le traitement est avantageux même s’il est entrepris après l’apparition des symptômes, mais certaines fonctions motrices s’acquérant tôt dans la vie ne peuvent être récupérées une fois qu’elles sont perdues.
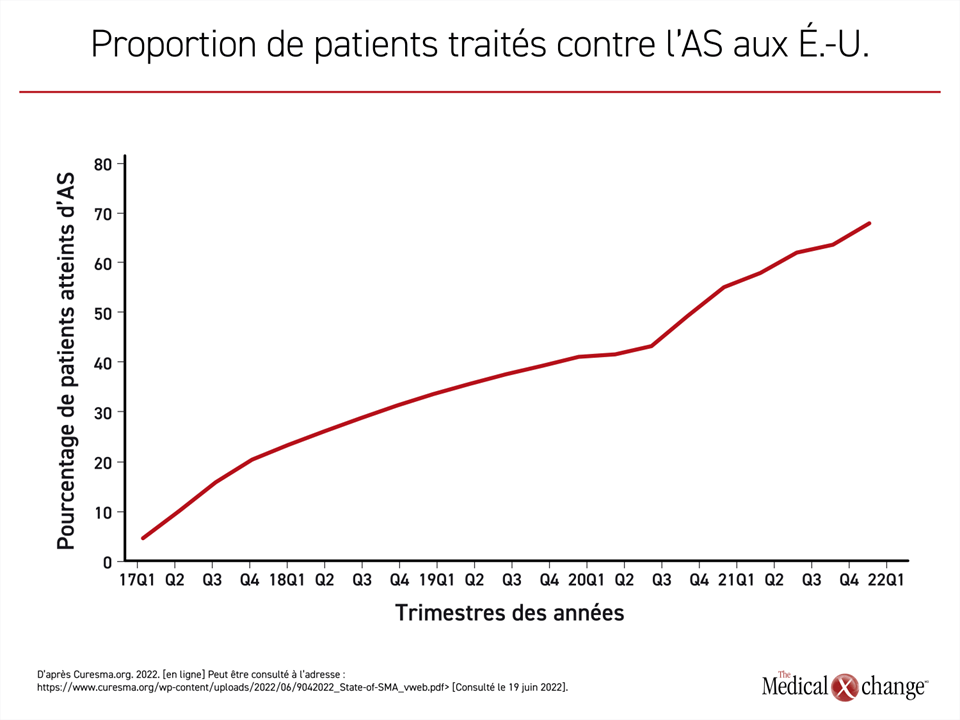
La courbe rendant compte de l’exposition grandissante au traitement est inversement proportionnelle à celle de la baisse des décès imputables à l’AS. En 2021, 97 % atteints d’AS à la naissance recevaient un médicament homologué, ce qui est en corrélation avec le nadir du taux de mortalité imputable à l’AS (Figure 2). En plus de son effet sur la mortalité, l’exposition croissante aux traitements correspond également à la proportion d’enfants atteints d’AS qui parviennent à s’asseoir sans aide. En 2021, le pourcentage des enfants capables de s’asseoir sans aide dépassait celui des enfants qui n’avaient pas franchi ce jalon pour une première fois (54 % vs 46 %).
Les traitements opposés à l’AS : les différences
L’amélioration des résultats obtenus depuis que des traitements sont disponibles n’a pas été stratifiée en fonction de chacun d’eux, mais ils comportent des différences. L’onasemnogène abéparvovec (OA) est un agent de thérapie génique; il remplace le gène SMN1 dysfonctionnel, ce qui permet de rétablir l’expression de la protéine SMN. Une seule dose suffit. Le nusinersen, le premier agent à avoir été homologué, s’administre par perfusion intrathécale et le risdiplam, dont l’homologation a suivi de peu celle de l’OA, se prend par voie orale.
Tout comme le nusinersen, le risdiplam stimule la production de protéine SMN par le gène SMN2. Ces deux agents compensent la production insuffisante de cette protéine, mais ils n’éliminent pas la cause de l’AS pour autant, sans compter qu’ils doivent être administrés indéfiniment pour que leurs effets positifs persistent.
Jusqu’à maintenant, l’OA offre une maîtrise durable de l’AS aux enfants qui y répondent. Contrairement au nusinersen et au risdiplam, cet agent de thérapie génique est indiqué jusqu’à l’âge de 24 mois. Bien que l’OA ait aussi des effets positifs chez les enfants symptomatiques, la thérapie génique doit idéalement être administrée tôt, puisqu’il se peut qu’une seule dose permette aux enfants de se développer normalement.
L’objectif principal : préserver le fonctionnement bulbaire
Les études cliniques ayant porté sur la surveillance du fonctionnement bulbaire dont dépendent les fonctions essentielles comme la respiration, la déglutition et la communication élémentaire, ont témoigné de l’utilité de l’OA pour préserver et rétablir le développement neurologique fondamental. D’après la Dre Katlyn E. McGrattan, professeure adjointe à l’Université du Minnesota, à Minneapolis, les données issues de ces études révèlent que la majorité des enfants atteints d’AS traitée par la thérapie génique possèdent les aptitudes auxquelles on s’attend chez des enfants de leur âge.
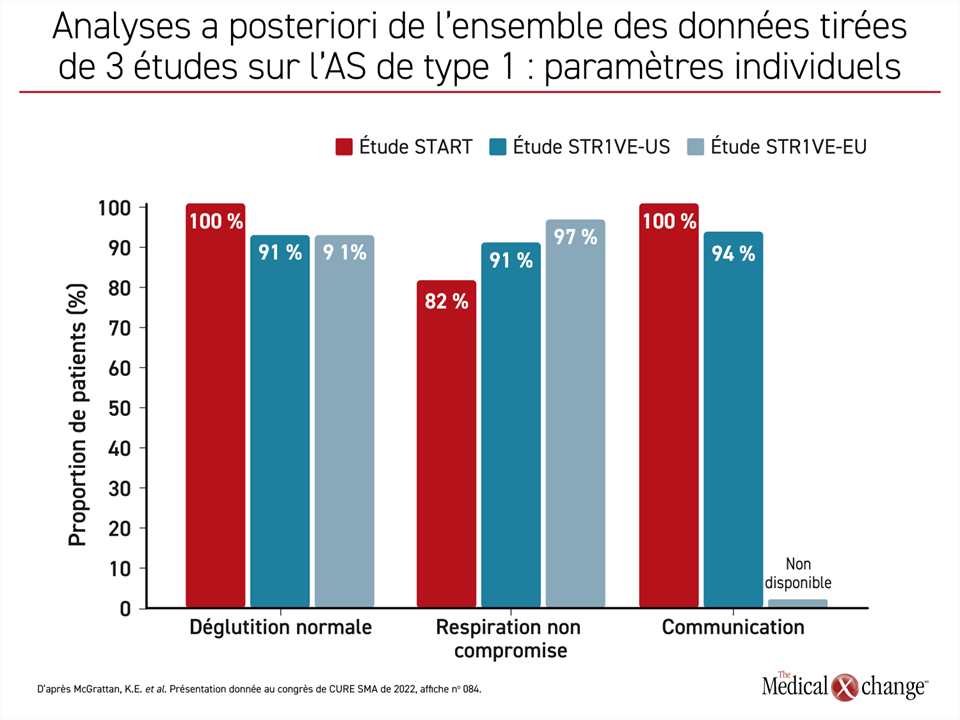
« Nous avons examiné le fonctionnement bulbaire de 54 enfants atteints d’AS ayant participé aux études de phase III STR1VE-US et STR1VE-EU et celle de 11 enfants ayant participé à l’étude de phase I START », a expliqué la Dre McGrattan. Ils ont tous été traités avant l’âge de 6 mois. Les patients des études de phase III ont été suivis pendant 18 mois, et ceux de l’étude de phase I, pendant 24 mois.
La Dre McGrattan a affirmé que l’évaluation des fonctions principales avait révélé qu’« à la fin de la période de suivi, 95 % des enfants satisfaisaient aux critères établis pour la communication, la déglutition et la respiration ». Plus précisément, elle a parlé de communication suffisamment compréhensible pour un inconnu, d’une déglutition fonctionnelle et de la satisfaction des besoins nutritionnels sans assistance respiratoire et sans incident respiratoire grave (Figure 3).
« À la fin de la période de suivi, 95 % des enfants satisfaisaient aux critères établis pour la communication, la déglutition et la respiration. »
Nul ne pouvait s’attendre à ce que ces patients, qui étaient tous atteints de la forme grave d’AS, franchissent le cap des 18 mois de vie tout en préservant leur fonctionnement bulbaire. Or plusieurs études sur la thérapie génique ont servi à évaluer des jalons beaucoup plus difficilement atteignables et d’un niveau fonctionnel plus élevé.
L’étude SPR1NT : franchissement des jalons malgré une maladie autrement mortelle
L’une de ces nombreuses études, l’étude de phase III SPR1NT, dont le recrutement était réservé aux patients porteurs de 3 copies du gène SMN2, avait pour objectif de vérifier si l’OA permettait de prévenir les symptômes et non pas seulement de les retarder. Même si ces patients parviennent souvent à s’asseoir seuls sans traitement, ils ne réussissent habituellement jamais à marcher et à se tenir debout sans aide.
Lors de l’étude SPR1NT, « les 15 enfants (100 %) ont tous réussi à se tenir debout sans aide, soit le paramètre d’évaluation principal de l’efficacité », a rapporté le Dr Kevin Strauss, directeur médical de la Clinic for Special Children, de Strasburg, en Pennsylvanie.
« Avec la thérapie génique, les 15 enfants (100 %) ont tous réussi à se tenir debout sans aide, soit le paramètre d’évaluation principal de l’efficacité. »
Une épreuve de dépistage réalisée à la naissance a permis de diagnostiquer l’AS de la plupart des sujets de l’étude SPR1NT. Le traitement par l’OA a été amorcé dans les semaines qui ont suivi leur naissance. Comme il fallait s’y attendre, tous les enfants affichent un fonctionnement bulbaire standard et la majorité d’entre eux ont un développement normal pour leur âge, et ce pour tous les paramètres de mesure ou presque.
« Onze enfants sont capables de s’asseoir seuls, 14 réussissent à se tenir debout sans appui et 11 ont commencé à marcher sans aide à un âge jugé normal. »
Par exemple, onze enfants sont capables de s’asseoir seuls, 14 réussissent à se tenir debout sans appui et 11 ont commencé à marcher sans aide à un âge jugé normal. La majorité des autres y sont parvenus avec un peu de retard. Au bout de 24 mois au moins de suivi, aucun des enfants n’avait régressé. Il faut souligner en outre qu’aucun des enfants n’a eu besoin d’assistance respiratoire ni de la moindre sonde d’alimentation pendant l’étude, ce qui est en contradiction avec l’évolution naturelle de la maladie.
Durée prometteuse de la réponse à la thérapie génique
Nous n’avons pas assez de recul pour conclure que la thérapie génique permettra de préserver le fonctionnement moteur indéfiniment, mais selon le Dr Strauss, certains des enfants ayant participé aux études de phase I bénéficient des gains réalisés à ce chapitre depuis plus de 5 ans.
Certains effets indésirables ont été observés. Quatre (26 %) des 15 participants à l’étude SPR1NT ont montré des signes d’altération de leur fonction hépatique, de grade 3 pour l’un d’eux. Cela dit, ils se sont tous rétablis, qu’ils aient été traités (p. ex., avec une corticothérapie) ou non. De plus, 2 (13 %) patients ont eu des problèmes liés à une thrombopénie, mais les examens effectués par la suite, par échographie notamment, n’ont fait ressortir aucune manifestation clinique d’importance ni d’anomalies cardiologiques.
« Un enfant a éprouvé deux effets indésirables possiblement liés à une ganglionite, mais le clinicien-chercheur a jugé qu’aucun d’eux n’avait de lien avec l’OA », a affirmé le Dr Strauss, qui a précisé qu’aucun de ces effets indésirables n’a laissé de séquelles graves.
Tous les traitements administrés avant l’apparition des symptômes ont été avantageux
Le nusinersen et le risdiplam se sont eux aussi montrés efficaces contre l’AS lorsqu’ils étaient administrés avant l’apparition des symptômes. Bien que plusieurs des études initiales menées sur ces agents l’aient été chez des enfants plus vieux, dont certains étaient atteints d’une forme plus légère et symptomatique de la maladie, les études réalisées chez des enfants présymptomatiques confirment l’intérêt de la prévention des déficits neurologiques fonctionnels. Au cours de l’étude NURTURE, une étude ouverte de phase II multicentrique sur le nusinersen, 22 (88 %) des 25 enfants marchaient sans aide au terme d’un suivi de 2,9 ans (De Vivo, D.C. et al. Neuromusc Disord. 2019;29:842-856).
Selon les données de l’étude RAINBOWFISH présentée lors de la conférence clinique et scientifique de 2022 de la Muscular Dystrophy Association (MDA) (en attente de publication), la plupart des nourrissons de moins de 2 mois présymptomatiques y ayant participé ont réussi à asseoir sans aide. Toutes les données du suivi sur les autres jalons n’avaient pas encore été recueillies, mais la FDA a tout de même homologué l’emploi du risdiplam chez les nourrissons à la lumière des résultats de l’étude RAINBOWFISH.
Les trois agents offerts pour le traitement de l’AS n’ont pas été comparés les uns aux autres, mais le nusinersen et le risdiplam exigent une administration chronique et une bonne observance thérapeutique. On ignore quels peuvent être les risques d’un maintien rigoureux de concentrations appropriées de protéine SMN, mais on sait qu’il est difficile de conserver une bonne observance thérapeutique la vie durant. Le nusinersen doit être administré par perfusion intrathécale tous les quatre mois après les doses d’attaque. Comme il est administré par voie orale, le risdiplam permet d’éviter les perfusions périodiques, mais les patients ont du mal à être disciplinés et persévérants lorsqu’ils doivent prendre des médicaments par voie orale, un fait bien établi dans la littérature médicale.
Les répercussions de l’AS chez les proches aidants
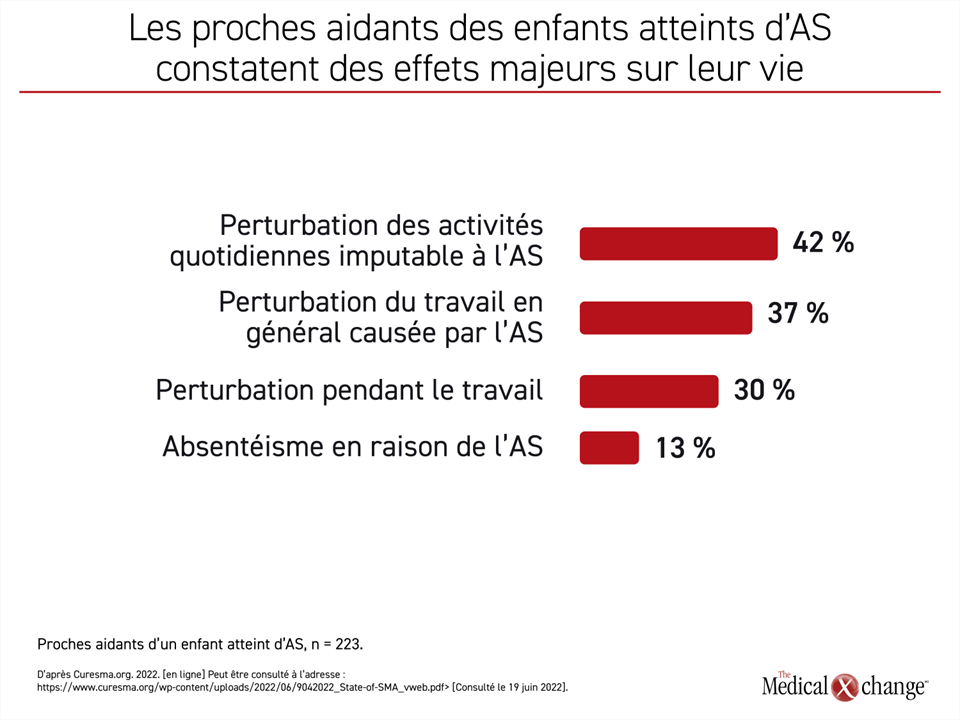
Le traitement de l’AS n’a pas seulement amélioré considérablement le pronostic des patients nés avec cette maladie. La meilleure maîtrise de la maladie qu’il permet d’obtenir a probablement aussi un retentissement sur la vie des proches aidants. Les auteurs du rapport State of SMA publié récemment n’ont pas évalué les répercussions des agents offerts aujourd’hui contre l’AS pour les proches aidants, mais ils y ont résumé les résultats d’une enquête montrant l’importance du fardeau que représente la maladie. Les données recueillies auprès des proches aidants n’ont pas été stratifiées en fonction de la gravité de l’AS, mais elles nous apprennent que 14 % d’entre eux disent s’être absentés de leur travail à l’occasion, 37 % ont rapporté que leur travail avait souffert de la maladie de leur enfant et 42 % ont affirmé qu’elle avait nui à leurs activités quotidiennes (Figure 4).
Conclusion
Le premier rapport annuel State of SMA, qui a été remis aux participants au congrès annuel de 2022 sur la recherche sur l’AS et les soins cliniques opposés à cette maladie, confirme que le sort des patients s’est grandement amélioré, notamment grâce à une baisse considérable des décès, depuis 2017, année où ont été lancés des agents opposés à cette maladie. La pente descendante de la courbe de la mortalité, qui semble poursuivre sur sa lancée, s’est révélée inversement proportionnelle à la courbe ascendante de l’exposition à la thérapie génique. Le plein effet du traitement sur le pronostic à long terme de l’AS devra faire l’objet d’un suivi supplémentaire, mais il demeure que les traitements entrepris avant l’apparition des symptômes semblent permettre à beaucoup d’enfants de franchir normalement les différentes étapes de leur développement. Or les données sur la thérapie génique indiquent qu’il est possible d’obtenir pareil résultat avec un seul traitement.