Neurology
2022 Annual SMA Research & Clinical Care Meeting
State of SMA Report Confirms Large Treatment-Related Improvements in Spinal Muscular Atrophy Outcomes
Anaheim – There has been a large reduction in the mortality rate among patients with spinal muscular atrophy (SMA) since therapies were introduced 5 years ago, according to an analysis of contemporary data. Published in a document called State of SMA distributed to those attending the 2022 Annual SMA Research & Clinical Care Meeting, this analysis has found that the improvement in outcomes, including survival, extends to disease severities that had been uniformly fatal. Post-hoc analyses of pooled data from three studies on preserving and stabilizing bulbar function, and final data from the SPR1NT study detailing major benefits with gene replacement therapy in pre-symptomatic infants, were also reported here.
The efficacy of the three available treatments for SMA derives from their ability to reverse survival motor neuron (SMN) protein deficiency, the cause of the disease. Two require maintenance treatment, while a gene replacement therapy restores production of the protein. The first therapy, requiring repeated intrathecal doses, was approved 5 years ago. The single-dose gene replacement therapy and a daily oral therapy followed.
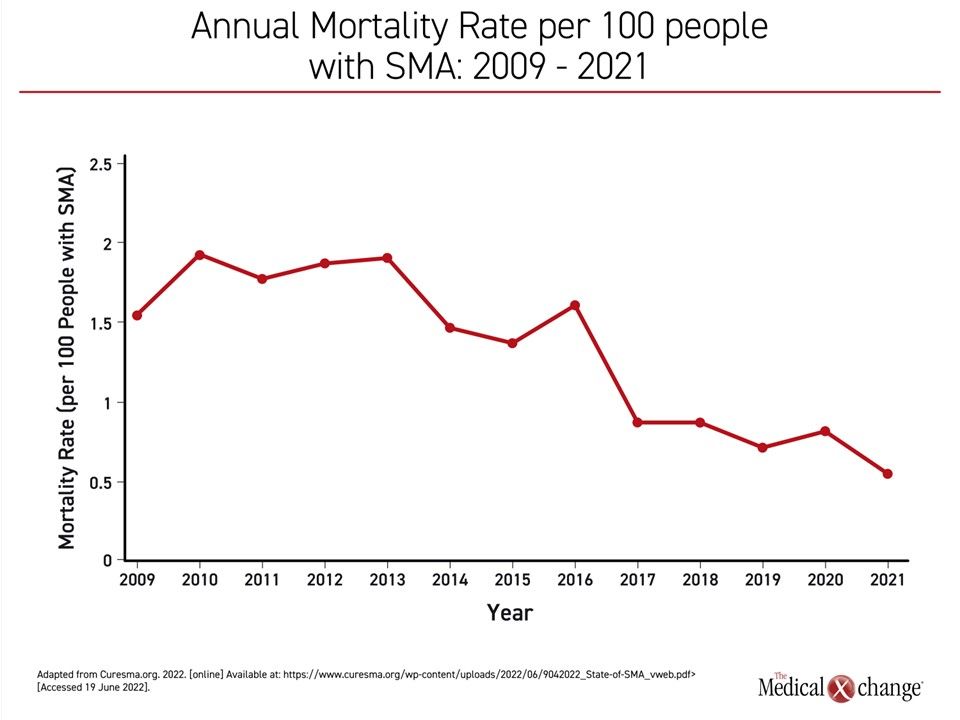
From the introduction of treatment, deaths per 100 patients with SMA have been reduced almost 75%, falling from an annual rate of nearly 2.0 per 100 patients a decade ago to 0.5 in 2021 (Figure 1). Favorable changes in other outcomes, including disability, have been on the same scale, according to the authors of the report, including Dr. Mary Schroth, Chief Medical Officer, Cure SMA, and a team of data analysts.
Further SMA Mortality Reductions Expected
The improvement in outcomes is likely to continue. Rapidly growing rates of newborn screening will increase intervention earlier in life. “Increased newborn screening has been accompanied by a decreasing frequency of symptomatic diagnosis,” explained Dr. Schroth and coauthors. By screening and then treating children prior to the onset of symptoms, irreversible neurofunctional deficits are avoided.
All treatments increase availability of the deficient SMN protein. While a minority of patients with SMA have a milder form, most have a severe disease defined by a low number of SMN2 gene copies. For these patients, survival is less than 2 years without treatment. Treatment is beneficial even following the onset of symptoms, but some motor functions developed early in life cannot be regained once lost.
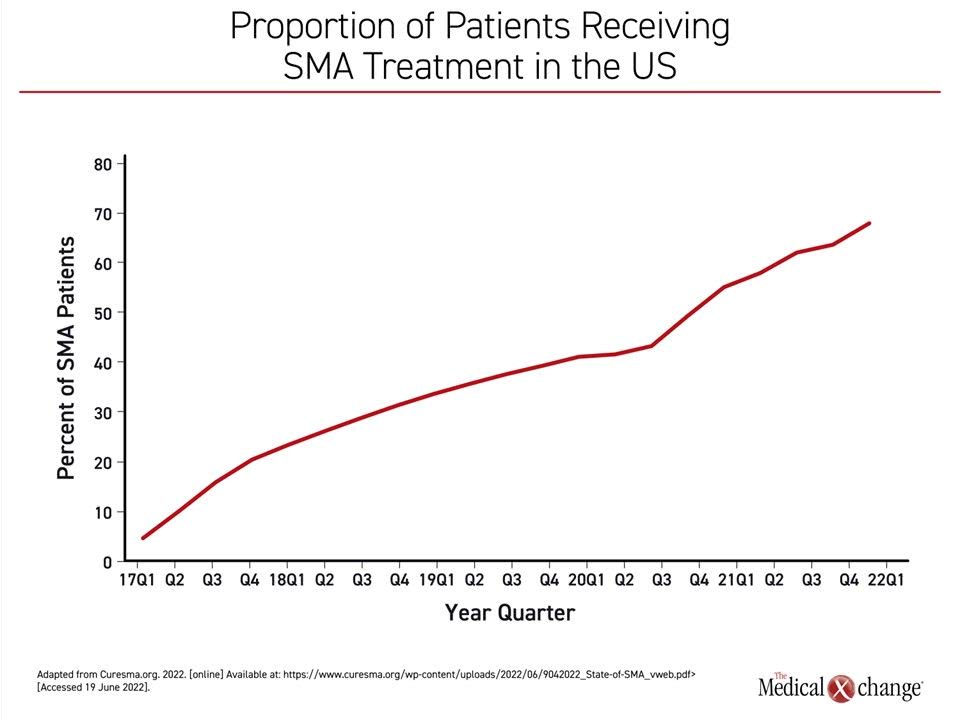
The curve showing increasing exposure to treatment is inversely related to the curve showing reductions in SMA-related death. By 2021, 97% of children born with SMA were receiving an approved therapy, correlating with the nadir in the rate of SMA mortality (Figure 2). In addition to the impact on mortality, the rising rates of exposure to treatment also correspond to the proportion of children with SMA sitting without support. In 2021, the proportion sitting without support exceeded the proportion who has not achieved this milestone for the first time (54% vs. 46%).
SMA Therapies: The Differences
The improvement in outcomes associated with the availability of treatment has not been stratified by type of treatment, but these differ. Onasemnogene abeparvovec (OA) is a gene replacement therapy that replaces the dysfunctional SMN1 gene to restore SMN protein expression. No further therapy is needed after initial administration. Nusinersen, the first approved therapy, is delivered by intrathecal infusion. Risdiplam, approved soon after OA, is an oral therapy.
Like nusinersen, the benefit of risdiplam is derived from stimulating SMN protein production from the SMN2 gene. They compensate for deficient SMN production but do not directly address the cause of SMA. To preserve benefit, nusinersen and risdiplam are maintained indefinitely.
OA so far provides indefinite control of SMA in responding children. The indication for this gene replacement therapy, unlike nusinersen or risdiplam, is for children up to 24 months of age. Although OA is associated with benefit in symptomatic children, early administration of gene replacement therapy is the goal for its potential to provide age-appropriate development after a single dose.
Primary Goal: Maintain Bulbar Function
The value of OA in preserving and restoring fundamental neurodevelopment is reflected in clinical studies that have monitored bulbar function, which describes fundamental functions, such as respiration, swallowing, and basic communication. The data from these studies show appropriate-for-age abilities in most children with SMA who receive the gene replacement therapy, according to Dr. Katlyn E. McGrattan, Assistant Professor, University of Minnesota, Minneapolis.
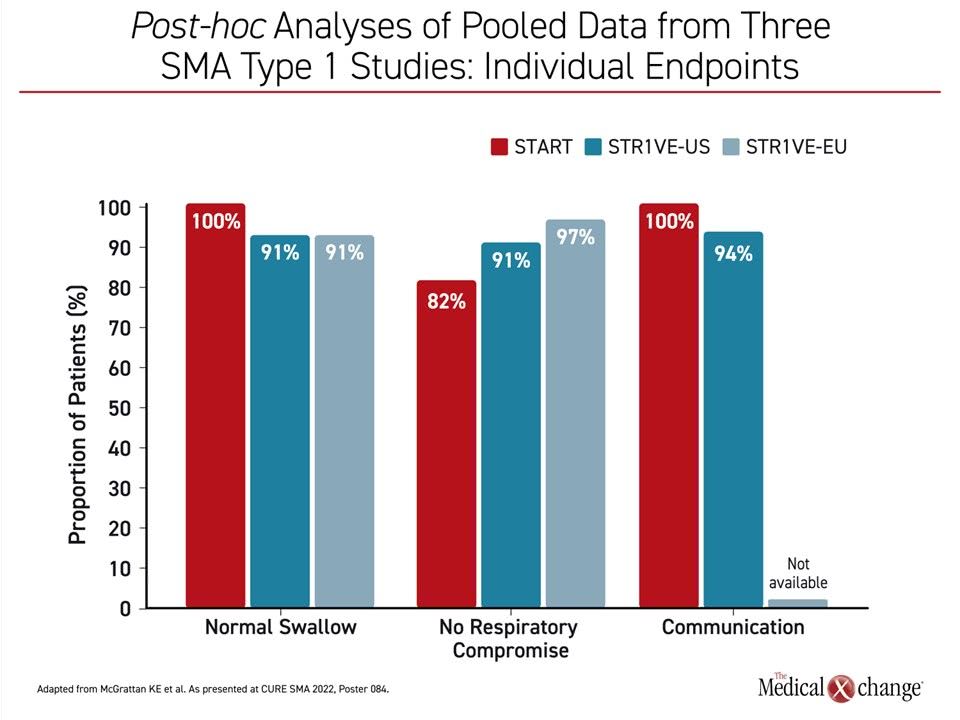
“We looked at bulbar function across 54 children with SMA who participated in the phase 3 trials STR1VE-US and STR1VE-EU as well as 11 children who were in the phase 1 START trial,” Dr. McGrattan explained. All were treated before six months of age. Patients in the phase 3 trials were followed for 18 months and those in the phase 1 study were followed for 24 months.
In those patients assessed for key functions, “95% met the communication, swallowing and respiratory endpoints by the end of follow-up,” according to Dr. McGrattan who specified these as, respectively, communicate sufficiently with comprehension by an unknown listener, functional swallow, and meeting nutritional needs with unassisted breathing and no serious respiratory events (Figure 3).
In these patients, all of whom had the severe form of SMA, none would have been expected to reach 18 months of life with preservation of bulbar function, but several of the studies with gene replacement therapy have monitored much more challenging milestones associated with a greater level of function.
SPR1NT Study: Milestones Reached in Otherwise Fatal Condition
Of several such studies, the phase 3 SPR1NT study, which restricted enrollment to patients with 3 SMN2 copies, the goal was to evaluate whether OA could prevent, not just delay symptoms. Although SMA children with 3 SMN2 copies often achieve the ability to sit independently without treatment, they do not typically ever achieve the ability to stand or walk independently.
In the SPR1NT study, “all 15 children—100%—achieved the primary efficacy endpoint of stands alone,” reported Dr. Kevin Strauss, Medical Director, Clinic for Special Children, Strasburg, Pennsylvania.
SMA was diagnosed with newborn screening in most SPR1NT patients. OA treatment was started within weeks of birth. Not surprisingly, all children achieved standard bulbar function, but most of these patients are developing within the expected age-appropriate developmental window across all or most measures.
For example, 11 of the children achieved independent sitting, 14 achieved independent standing, and 11 are walking independently within the age-appropriate developmental window. Most of those who did not reach the milestone did so with delay. After at least 24 months of follow-up there has been no loss of the developmental milestones that were gained earlier. Of particular interest, all children were free of any ventilatory or feeding tube support of any kind during the study, in sharp contrast to the natural progression of this disease.
Duration of Response to Gene Replacement Therapy Promising
Follow-up is not yet long enough to conclude that gene replacement therapy will preserve motor function indefinitely, but there are patients who participated in the phase 1 studies who have preserved their gains in motor function for more than 5 years, according to Dr. Strauss.
There have been some noted side effects. Four (26%) of the 15 participants in the SPR1NT trial have had signs of impaired hepatic function, including one of grade 3 severity. However, all have self-resolved or resolved with treatment, such as steroids. In addition, 2 (13%) patients have had thrombocytopenia-related events even though further workup, including echocardiograms, did not substantiate clinical events or cardiological abnormalities.
“Two potential ganglionitis-related adverse events occurred in one child, but neither was considered to be related to OA by the investigator,” said Dr. Strauss, who noted that none of these side effects have been associated with serious sequelae.
All Treatments Given Pre-symptomatically Are Beneficial
Nusinersen and risdiplam have also demonstrated efficacy against SMA when initiated pre-symptomatically. Although many of the initial studies with these agents were conducted in older children, including those with symptomatic milder forms of SMA, the studies in pre-symptomatic children support the value of preventing neurofunctional deficits. In the phase 2 open-label multicenter NURTURE trial with nusinersen, 22 (88%) of 25 children achieved independent walking in a follow-up of 2.9 years (De Vivo DC et al. Neuromusc Disord. 2019;29:842-856).
In the as-yet unpublished RAINBOWFISH study, which included pre-symptomatic infants less than 2 months of age, sitting without support was achieved in most patients, according to data presented at the 2022 Muscular Dystrophy Association (MDA) Clinical & Scientific Conference. Follow-up data were not yet complete on other milestones, but the FDA approved risdiplam for infants on the basis of RAINBOWFISH.
The three available therapies for SMA have not been compared directly, but nusinersen and risdiplam are dependent on indefinite treatment and compliance. The risks from rigorously maintaining adequate levels of the SMN protein are unknown, however adherence over a lifetime may prove challenging. For intrathecal nusinersen, doses are required every four months after a loading regimen. For risdiplam, oral dosing avoids this periodic infusion, but the difficulty of maintaining high levels of adherence over extended periods of time to oral therapy are well documented in medical literature.
Considering the Impact of SMA on Caregivers
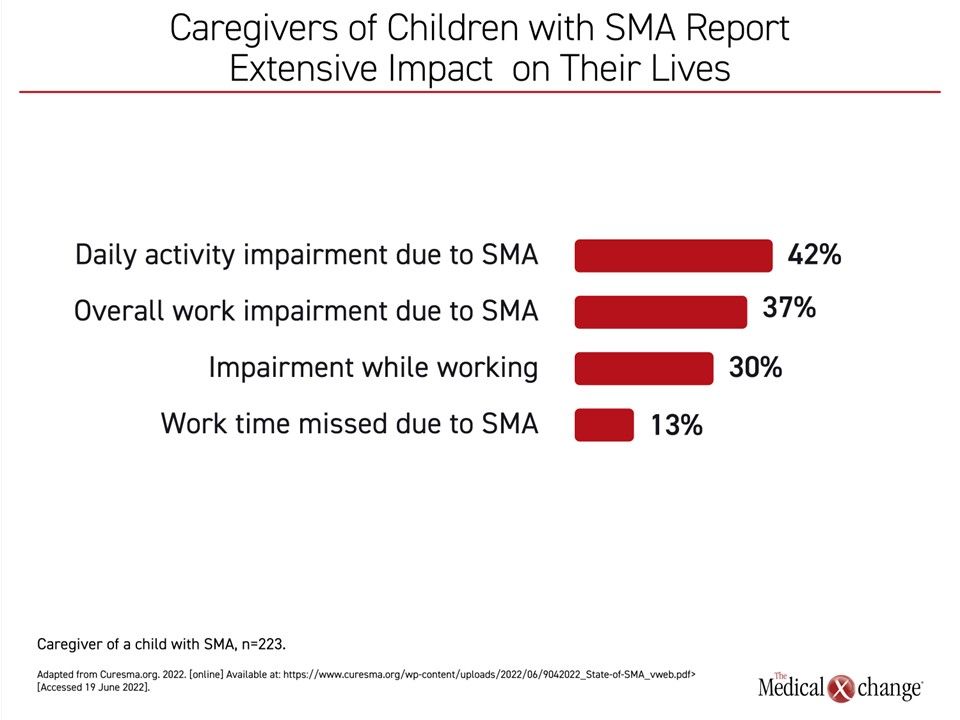
Treatment of SMA has not only meant major improvements in the prognosis of patients born with SMA, but the improvements in disease control are likely to impact caregivers. The newly released State of SMA report did not evaluate the impact of the availability of treatments for SMA on the caregiver experience, but it did summarize a survey that shows the burden of disease is substantial. Without breaking down caregiver experience relative to disease severity, the respondent data showed that 14% occasionally miss work, 37% report work impairment related to their child’s disease, and 42% report impairment of daily life activities (Figure 4).
Conclusion
In the first annual State of SMA report distributed at the 2022 Annual SMA Research & Clinical Care Meeting, large improvements in outcomes, including major reductions in mortality, were documented following the introduction of SMA treatments in 2017. The downward slope of the mortality curve, which appears to be ongoing, has been inversely related to the upward slope of exposure to gene therapy. The full impact of treatment on the long-term prognosis of SMA requires additional follow-up, but therapies initiated before symptoms appear to permit many children to achieve the expected age-appropriate developmental milestones. Data with gene replacement therapy suggests this can be achieved with a single treatment.