Neurologie
ATTRh-PN : revue d'expert et commentaires tirés de la littérature
Nouvelles options de traitement contre l’amylose héréditaire à transthyrétine accompagnée de polyneuropathie
Geneviève Matte, MDCM, FRCPC
Neurologue, Centre hospitalier de l’Université de Montréal (CHUM)
Professeure adjointe de clinique, Département de neurosciences, Université de Montréal
Directrice, Clinique de la SLA et maladies du neurone moteur, CHUM
Montréal (Québec)
Vera Bril, B.Sc., M.D., FRCPC
Professeure de neurologie, Université de Toronto
Neurologue et directrice de la section neuromusculaire, Réseau de santé universitaire et Université de Toronto
Chercheuse principale, Institut de recherche de l’hôpital général de Toronto
Toronto (Ontario)
Les médicaments qui répriment l’expression du gène TTR codant pour la transthyrétine modifient l’évolution naturelle de l’amylose héréditaire à transthyrétine accompagnée de polyneuropathie (hATTR-PN). Administrés par injection ou par perfusion, ils inhibent la production de protéine TTR qui, lorsqu’elle est produite par un gène TTR muté, se replie anormalement et s’agrège dans les nerfs des systèmes périphérique et autonome, ce qui entrave leur fonctionnement. Ce mécanisme s’applique à d’autres sous-types de l’ATTR qui sont définis d’après leurs principales caractéristiques cliniques et qui, à l’instar de l’hATTR-PN, touchent plusieurs systèmes et appareils. L’étude de phase III NEURO-TTRansform a permis d’établir un lien entre le plus récent de ces agents et des baisses considérables des concentrations sériques de protéine TTR, une atténuation de l’atteinte neurologique et une meilleure qualité de vie. La possibilité que ce médicament réprimant l’expression du gène TTR, et d’autres comme lui, prolongent la survie continue d’être suivie de près.
Contexte
Le terme amylose regroupe un éventail hétérogène de troubles génétiques ou acquis entraînant l’agrégation de protéines fibrillaires à l’intérieur des tissus cibles et provoquant leur dysfonctionnement1. Contrairement à l’amylose à chaînes légères, la forme la plus répandue de ce trouble2, l’ATTR peut être une conséquence du vieillissement (forme sauvage) ou de mutations du gène TTR transmises génétiquement selon un mode autosomique dominant (forme héréditaire)3. Les sous-types les plus répandus sont l’ATTRh accompagnée de polyneuropathie (ATTRh-PN), qui se caractérise par l’accumulation de dépôts amyloïdes dans les nerfs des systèmes autonome et périphérique4, et l’ATTR accompagnée de myocardiopathie (ATTR-CM), où les dépôts amyloïdes s’accumulent dans le cœur5. Définis par leurs caractéristiques cliniques prépondérantes, l’évolution et le dysfonctionnement potentiellement mortel survenant chez les patients atteints de l’un ou l’autre sous-type peuvent toucher plusieurs appareils et organes6.
Pour une entrevue exclusive avec la Dre Geneviève Matte couvrant l’impact sur la pratique clinique, cliquez ici
Auparavant mieux connue sous l’appellation de polyneuropathie amyloïde familiale, l’ATTRh-PN, qui a été décrite pour la première fois en 19527, était considérée comme rare ou peu fréquente hors des pays où elle était à l’état endémique, tels que le Portugal, où les premiers cas ont été découverts7. Selon une estimation tirée d’une enquête réalisée en 2016, la prévalence de l’ATTRh-PN au Portugal se chiffrait à 23 cas par tranche de 100 000 adultes8. On ignore si ce chiffre est représentatif de ce qui se passe à l’échelle internationale, mais on a recensé des cas partout dans le monde9.
La grande hétérogénéité du tableau clinique de l’ATTRh-PN et de l’ATTR-CM, notamment leurs symptômes et leur gravité, s’explique surtout par les différentes mutations sous-jacentes du gène TTR. Selon une analyse récente, plus de 150 mutations de ce gène ont été répertoriées dans la base de données sur l’amylose héréditaire (Hereditary Amyloidosis Database)4. Pour ce qui est de l’ATTRh-PN, la première mutation du gène TTR découverte, Val30Met, est aussi la plus répandue, puisqu’elle est incriminée dans environ 70 % des cas observés dans les régions où la maladie est endémique6. Les anomalies du gène TTR associées à l’ATTR sont majoritairement des variants faux-sens produits par des mutations ponctuelles.
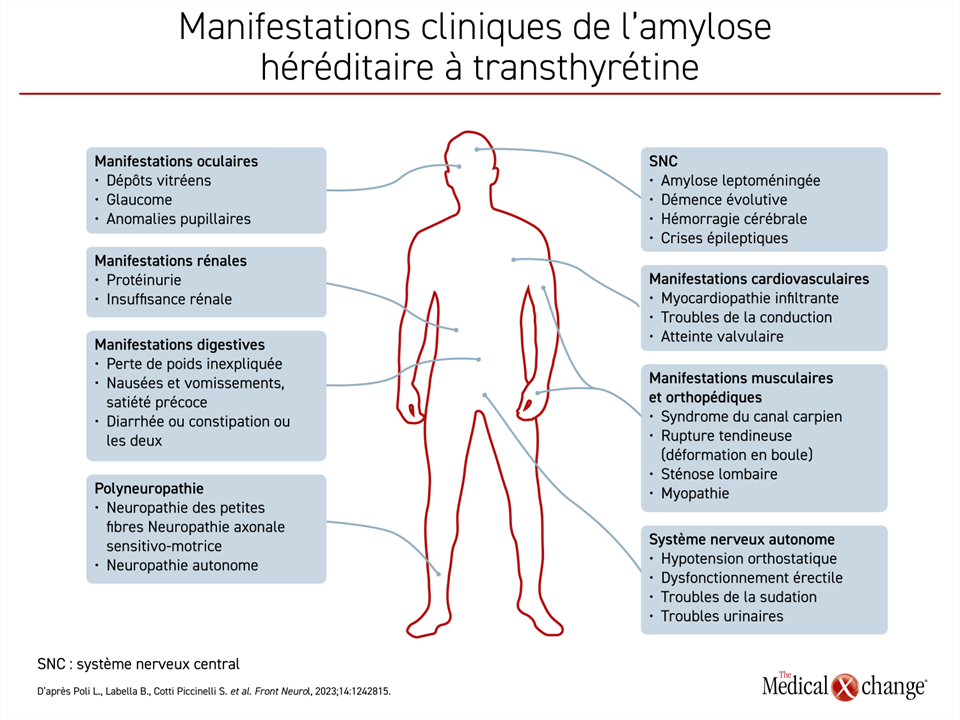
Hormis les effets nocifs exercés sur le système nerveux périphérique ou autonome et sur le cœur, l’ATTR peut entraîner un dysfonctionnement potentiellement mortel de divers autres systèmes et appareils tels que les reins, les yeux et le tube digestif (Figure 1). Le risque est établi d’après la mutation sous-jacente du gène TTR, du moins en partie. Exemple : le tiers environ des patients porteurs de la mutation Val30Met verront leur fonction rénale se dégrader peu à peu10.
Les techniques d’imagerie médicale, comme l’échographie, la tomographie assistée par ordinateur et l’imagerie par résonance magnétique (IRM), peuvent être utilisées pour évaluer l’atteinte des nerfs périphériques et du cœur, quoique l’évaluation des symptômes et de leur intensité soit une méthode souvent employée pour déterminer le fardeau pathologique et l’étendue de la maladie.
Traitement
Compte tenu de l’expression surtout hépatique de la protéine TTR, on a eu recours à la transplantation du foie pendant des décennies pour prolonger la survie des patients atteints d’ATTR, mais cette intervention n’est plus préconisée en première intention dans les lignes directrices canadiennes de 202211. Même si elle permet de prolonger la survie12, plusieurs problèmes comme les dépôts ininterrompus de substance amyloïde dans le cœur par la suite, font de cette opération13 une méthode inférieure aux traitements pharmacologiques offerts aujourd’hui.
L’arrivée sur le marché des agents servant à stabiliser la protéine TTR remonte à plus de 15 ans, mais ces derniers ont surtout été utilisés pour traiter l’ATTR-CM. En empêchant la dégradation de la protéine TTR, le diflunisal et le tafamidis, les deux agents de ce type les plus souvent employés, inhibent une étape essentielle de la formation des dépôts amyloïdes14,15. Le diflunisal n’a jamais été homologué par les organismes de réglementation pour le traitement de l’ATTR‑CM malgré les résultats positifs des études15,16. Le tafamidis a quant à lui été homologué contre l’ATTR-CM au Canada à la lumière des résultats d’une étude de phase III17, mais sa prescription est essentiellement réservée aux spécialistes. Aucun de ces deux agents n’est indiqué au pays pour traiter l’ATTRh-PN.
Au Canada, les médicaments réprimant l’expression du gène TTR ont été les premiers agents homologués expressément pour traiter l’ATTRh-PN. L’inotersen, un oligonucléotide antisens (OAS) de première génération, a ouvert le bal en 2018 suivi du patisiran, un petit ARN interférent (pARNi). L’inotersen, qui s’administre par injections sous-cutanées hebdomadaires, se lie à l’ARN messager du gène TTR (ARNm) pour inhiber la synthèse de la protéine TTR18, alors que le patisiran, qui s’administre par perfusion intraveineuse, est assimilé par les hépatocytes où il déclenche une dégradation catalytique de l’ARNm du gène TTR, ce qui provoque une baisse de la protéine TTR circulante19.
Pendant l’étude NEURO-TTR20 sur laquelle l’homologation de l’inotersen est fondée, 172 patients atteints d’ATTRh-PN ont été répartis aléatoirement selon un rapport de 2:1 de façon à recevoir des injections sous-cutanées hebdomadaires d’inotersen ou d’un placebo. Le paramètre d’évaluation principal était le score mNIS+7 (modified Neuropathy Impairment Score+7); une augmentation de ce score indique une détérioration des fonctions neurologiques. Or l’augmentation du score mNIS+7 observée chez les patients traités par l’agent réprimant l’expression du gène TTR a été significativement moindre que celle enregistrée chez les témoins (+5,8 vs +25,5 points; p < 0,001). Les patients du groupe inotersen ont aussi bénéficié d’une amélioration significativement plus grande de leur qualité de vie. Parmi les effets secondaires de l’inotersen, une glomérulonéphrite et une thrombopénie ont été mortelles chez un patient.
Lors de l’étude APOLLO sur laquelle repose l’homologation du patisiran, 225 patients atteints d’ATTRh‑PN ont été répartis aléatoirement selon un rapport de 2:1 de façon à recevoir toutes les 3 semaines des perfusions de patisiran ou d’un placebo21. La variation moyenne du paramètre d’évaluation principal, c’est-à-dire la variation du score mNIS+7, (-6,0 vs +28,0 points; p < 0,001) avantageait nettement le patisiran. Les paramètres d’évaluation secondaires, soit la vitesse de marche et la qualité de vie, se sont aussi améliorés significativement. Le patisiran a été bien toléré; il a toutefois provoqué plus de réactions au point d’injection que le placebo (20 % vs 10 %).
Le vutrisiran22, un autre pARNi dirigé contre l’ATTRh-PN, a été homologué par Santé Canada en 2023 sur la base d’une étude de phase III (HELIOS-A) pendant laquelle 164 patients ont été répartis aléatoirement selon un rapport de 3:1 de façon à recevoir des injections sous-cutanées de vutrisiran tous les 3 mois ou des perfusions intraveineuses de patisiran toutes les 3 semaines. Le groupe placebo de l’étude APOLLO a fait office de groupe témoin externe. La réduction de la concentration sérique de protéine TTR obtenue avec le vutrisiran n’était pas inférieure à celle observée avec le patisiran et le vutrisiran a été relativement bien toléré.
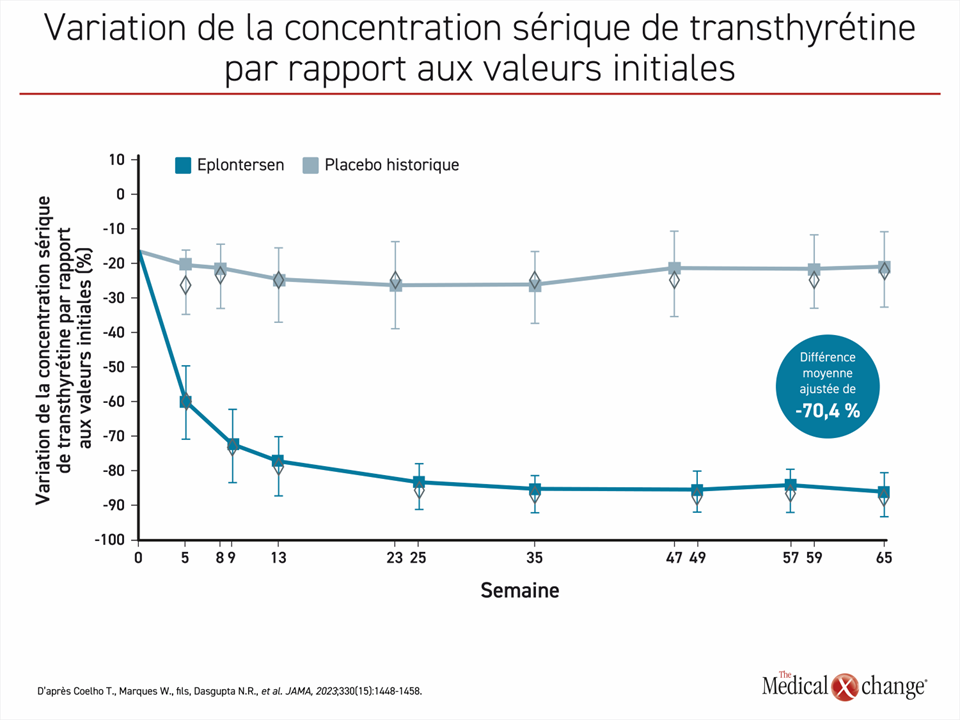
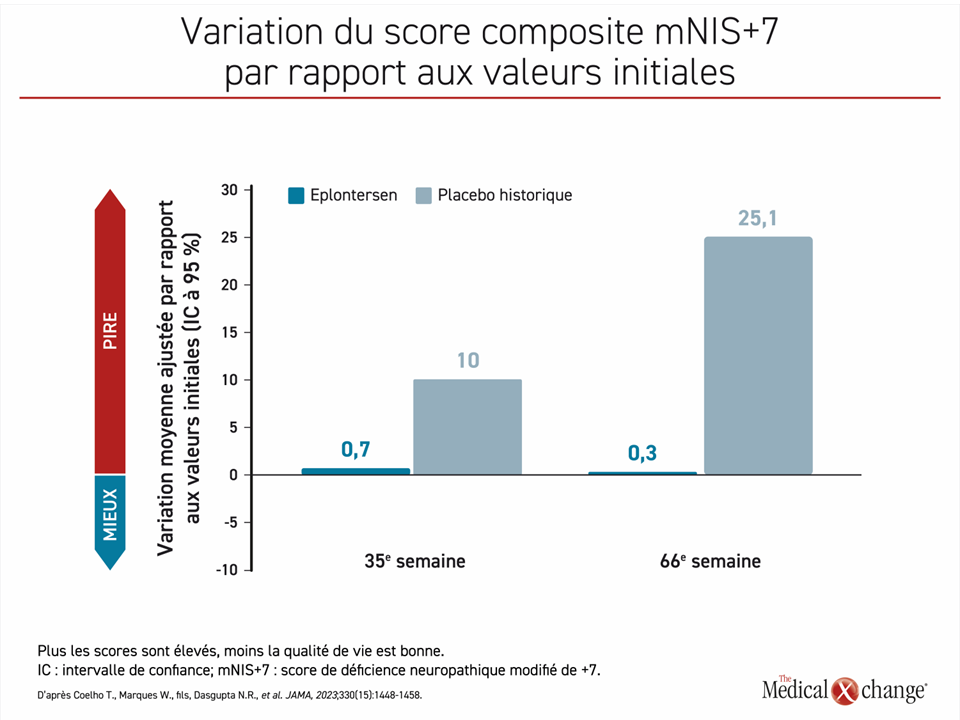
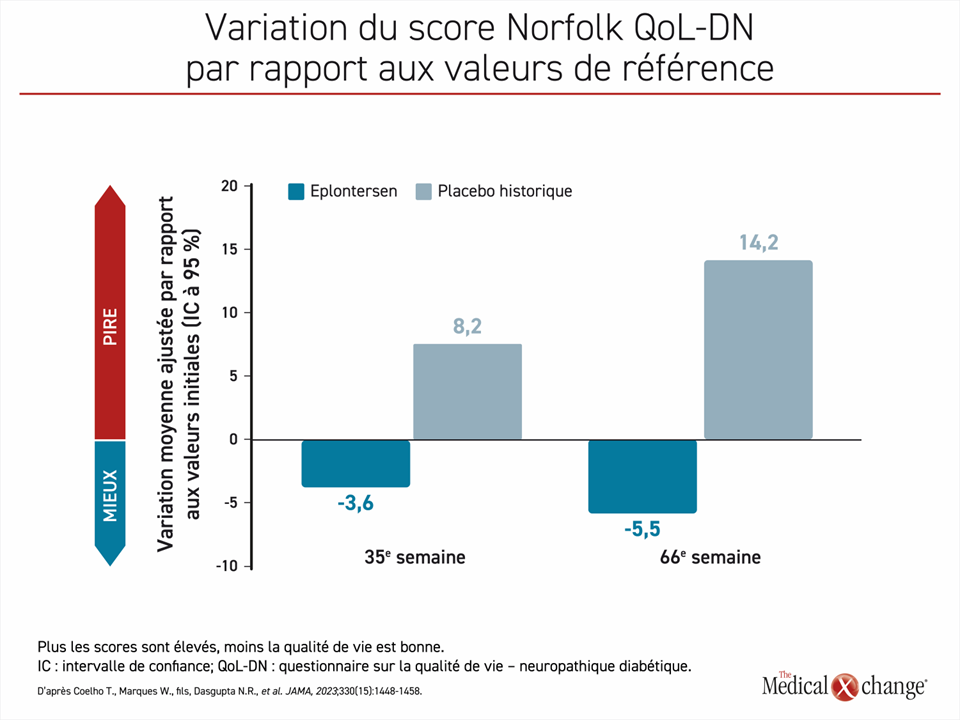
L’eplontersen est le plus récent des médicaments réprimant l’expression du gène TTR pour lequel une étude de phase III est terminée (NEURO-TTRansform)23. Lors de cette étude en mode ouvert, des patients ont été répartis aléatoirement selon un rapport de 6:1 de façon à recevoir de l’eplontersen ou un agent de référence, l’inotersen. Le groupe placebo historique de l’étude NEURO-TTR, dont les résultats ont déjà été publiés, a servi de groupe comparatif. Selon le plan de l’étude publié, les chercheurs ont choisi ce mode de fonctionnement parce qu’ils jugeaient que l’utilisation d’un groupe placebo était contraire à l’éthique étant donné la disponibilité d’autres traitements à l’efficacité établie. Les 24 patients du groupe inotersen, l’agent de référence, sont passés à l’eplontersen à la 34e semaine de l’étude. À la 65e semaine, l’avantage de l’eplontersen sur le placebo pour chacune des trois mesures composant le paramètre d’évaluation principal était extrêmement significatif sur le plan statistique. Voici les résultats : variation de la concentration sérique de transthyrétine (-81,7 % vs ‑11,2 %; p < 0,001), variation du score mNIS+7 (0,3 vs 25,1; p < 0,001) et variation du score au questionnaire Norfolk QoL-DN (Norfolk Quality of Life Questionnaire-Diabetic Neuropathy; une augmentation de ce score témoigne d’une détérioration de la qualité de vie) (-5,5 vs 14,2; p < 0,001) (Figures 2 à 4).
Le taux d’abandons du traitement motivés par un effet indésirable était similaire dans le groupe eplontersen et dans le groupe placebo (4 % vs 3 %). Aucun cas de glomérulonéphrite n’a été rapporté. Les cas de thrombopénie ꟷ tous bénins et n’ayant pas exigé de modification de la dose ni provoqué de saignements ꟷ sont survenus à la même fréquence dans les deux groupes, soit 2 %.
Cette étude ne visait pas à comparer directement l’eplontersen à l’inotersen, un agent de première génération, mais leur mode d’action différent pourrait expliquer la faible incidence d’effets indésirables et l’invariable efficacité de l’eplontersen. L’inotersen et l’eplontersen sont des oligonucléotides antisens, mais le mode d’action de ce dernier repose sur une technologie novatrice de conjugaison à un ligand. La liaison de la N-acétyl galactosamine (GaINAc) à un OAS favorise sa capture par les hépatocytes en interagissant avec les récepteurs de l’asialoglycoprotéine. Les patients peuvent s’autoadministrer l’eplontersen facilement à l’aide d’un auto-injecteur, et ce à des intervalles posologiques beaucoup plus longs que ceux de l’inotersen (toutes les 4 semaines vs toutes les semaines).
Enjeux
L’atténuation des symptômes et l’amélioration de la qualité de vie au cours de la période de suivi de plus de 1 année indiquent que les agents réprimant l’expression du gène TTR modifient l’évolution naturelle de l’ATTRh-PN. Ces médicaments n’ont pas été comparés les uns aux autres directement, mais les études menées sur chacun d’eux donnent à penser qu’ils pourraient ne pas être comparables. Ils se sont tous montrés plus efficaces qu’un placebo, mais la différence de réponse mesurée au moyen du score mNIS+7 variait et leurs tableaux d’effets indésirables sont différents.
Notons qu’il faudra disposer d’un suivi de plus longue durée pour savoir si tous les agents réprimant l’expression du gène TTR peuvent prolonger la survie. Un gain du côté de la survie pourrait venir de l’inhibition relative des dépôts de protéine dans de nombreux appareils et systèmes et non pas uniquement des neuropathies. La première étude de phase III (APOLLO-B) menée avec le patisiran, un agent réprimant l’expression du gène TTR, chez des patients atteints d’ATTR-CM a pris fin récemment. Or elle a révélé mis au jour une préservation fonctionnelle à la fin du 12e mois24. Les résultats d’une étude comparable réalisée avec l’eplontersen (CARDIO-TTRansform) devraient être connus bientôt.
Le plus grand avantage des agents réprimant l’expression du gène TTR pourrait être la possibilité d’amorcer le traitement dès le début de la maladie, ce qui, à terme, dépend de la reconnaissance rapide de l’ATTR. L’obtention de gains encore plus marqués, s’il en est, en alliant des médicaments servant à stabiliser la protéine TTR aux agents réprimant l’expression du gène TTR n’a pas encore été étudiée.
Résumé
Souvent accompagnée de symptômes débilitants, l’ATTRh-PN est une maladie évolutive dont l’issue est fatale si elle n’est pas maîtrisée. De plus en plus d’agents réprimant l’expression du gène TTR ont été reliés à un ralentissement de l’évolution de la maladie et à une meilleure qualité de vie. L’étude de phase III NEURO-TTRansform a confirmé l’efficacité de l’eplontersen, le plus récent des agents réprimant l’expression du gène TTR opposés à l’ATTR et OAS de nouvelle génération. Son auto-administration à l’aide d’un auto-injecteur et son intervalle posologique relativement long font de lui un choix pratique. Les d’agents réprimant l’expression du gène TTR constituent une avancée majeure dans le traitement de l’ATTRh-PN et se montrent prometteurs pour celui de l’ATTR‑CM. Les études de suivi de longue durée serviront à évaluer leur capacité à prévenir les effets nocifs de la maladie sur les organes vulnérables et à possiblement prolonger la survie.
Références
- Gertz MA et Dispenzieri A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A Systematic Review. JAMA 2020;324(1):79-89. DOI: 10.1001/jama.2020.5493.
- Zanwar S, Gertz MA et Muchtar E. Immunoglobulin Light Chain Amyloidosis: Diagnosis and Risk Assessment. J Natl Compr Canc Netw 2023;21(1):83-90. DOI: 10.6004/jnccn.2022.7077.
- Gertz MA, Benson MD, Dyck PJ, et al. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J Am Coll Cardiol 2015;66(21):2451-2466. DOI: 10.1016/j.jacc.2015.09.075.
- Poli L, Labella B, Cotti Piccinelli S, et al. Hereditary transthyretin amyloidosis: a comprehensive review with a focus on peripheral neuropathy. Front Neurol 2023;14:1242815. DOI: 10.3389/fneur.2023.1242815.
- Ruberg FL, Grogan M, Hanna M, Kelly JW et Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol 2019;73(22):2872-2891. DOI: 10.1016/j.jacc.2019.04.003.
- Luigetti M, Romano A, Di Paolantonio A, Bisogni G et Sabatelli M. Diagnosis and Treatment of Hereditary Transthyretin Amyloidosis (hATTR) Polyneuropathy: Current Perspectives on Improving Patient Care. Ther Clin Risk Manag 2020;16:109-123. DOI: 10.2147/TCRM.S219979.
- Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952;75(3):408-427. DOI: 10.1093/brain/75.3.408.
- Ines M, Coelho T, Conceicao I, Duarte-Ramos F, de Carvalho M et Costa J. Epidemiology of Transthyretin Familial Amyloid Polyneuropathy in Portugal: A Nationwide Study. Neuroepidemiology 2018;51(3-4):177-182. DOI: 10.1159/000490553.
- Adams D, Koike H, Slama M et Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 2019;15(7):387-404. DOI: 10.1038/s41582-019-0210-4.
- Lobato L et Rocha A. Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol 2012;7(8):1337-1346. DOI: 10.2215/CJN.08720811.
- Alcantara M, Mezei MM, Baker SK, Breiner A, Dhawan P et Fiander A. Canadian Guidelines for Hereditary Transthyretin Amyloidosis Polyneuropahty Management. Can J Neurol Sci 2022;49:7-18.
- Yamashita T, Ando Y, Okamoto S, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology 2012;78(9):637-643. DOI: 10.1212/WNL.0b013e318248df18.
- Yazaki M, Mitsuhashi S, Tokuda T, et al. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am J Transplant 2007;7(1):235-242. DOI: 10.1111/j.1600-6143.2006.01585.x.
- Coelho T, Merlini G, Bulawa CE, et al. Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis. Neurol Ther 2016;5(1):1-25. DOI: 10.1007/s40120-016-0040-x.
- Sekijima Y, Dendle MA et Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006;13(4):236-249. DOI: 10.1080/13506120600960882.
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013;310(24):2658-2667. DOI: 10.1001/jama.2013.283815.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018;379(11):1007-1016. DOI: 10.1056/NEJMoa1805689.
- Ackermann EJ, Guo S, Benson MD, et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid 2016;23(3):148-157. DOI: 10.1080/13506129.2016.1191458.
- Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 2013;369(9):819-829. DOI: 10.1056/NEJMoa1208760.
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379(1):22-31. DOI: 10.1056/NEJMoa1716793.
- Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379(1):11-21. DOI: 10.1056/NEJMoa1716153.
- Adams D, Tournev IL, Taylor MS, et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 2023;30(1):1-9. DOI: 10.1080/13506129.2022.2091985.
- Coelho T, Marques W, fils, Dasgupta NR, et al. Eplontersen for Hereditary Transthyretin Amyloidosis With Polyneuropathy. JAMA 2023;330(15):1448-1458. DOI: 10.1001/jama.2023.18688.
- Maurer MS, Kale P, Fontana M, et al. Patisiran Treatment in Patients with Transthyretin Cardiac Amyloidosis. N Engl J Med 2023;389(17):1553-1565. DOI: 10.1056/NEJMoa2300757.