Neurologie
27e congrès annuel international hybride de la World Muscle Society (WMS)
Amyotrophie spinale : la thérapie génique favorise un développement adapté à l’âge
Halifax – Deux séries de données montrent qu’une thérapie génique homologuée contre le traitement de l’amyotrophie spinale (AS) permet à la plupart des enfants de se développer normalement si elle est amorcée avant l’apparition des symptômes. Or il est souvent possible de ralentir considérablement l’évolution de la maladie même si elle l’est après. Ces observations ont d’abord été rapportées dans le cadre des études de phase III ayant mené à l’homologation d’une thérapie génique par les organismes de réglementation, mais les données d’un registre révèlent que les mêmes effets positifs sont obtenus en pratique clinique. Plus les données de suivi de ces deux séries s’accumulent, plus elles portent à croire qu’une fois qu’un enfant a franchi une étape de son développement, c’est pour longtemps, voire définitivement.
Avant l’arrivée relativement récente d’un traitement, les formes les plus graves d’AS étaient inexorablement mortelles, puisqu’elles emportaient généralement les enfants avant l’âge de 2 ans. Même si les enfants traités vivent plus longtemps aujourd’hui, il demeure que la thérapie génique à base d’onasemnogène abéparvovec (OA) a permis à beaucoup d’entre eux de se développer normalement et pas simplement de survivre. Bien que l’OA ait été homologué pour le traitement des enfants pesant moins de 21 kg à la lumière des effets positifs qu’il a exercés durant les études menées aux fins d’homologation, il faut savoir que des enfants âgés de quelques semaines à peine ont été recrutés dans les études de phase III. Notons que la plupart de ces derniers ont franchi normalement les étapes de leur développement, certains d’entre eux étant suivis depuis plusieurs années.
Pour une entrevue exclusive avec le Dr Laurent Servais couvrant l’impact sur la pratique clinique, cliquez ici.
L’AS se caractérise par un déficit en protéine SMN causé par un dysfonctionnement du gène SMN1. Cette protéine est essentielle aux motoneurones qui régissent le fonctionnement bulbaire et beaucoup d’autres fonctions motrices. Le gène SMN2 peut produire une certaine quantité de protéine SMN pour compenser, mais pas suffisamment. Le nusinersen et le risdiplam, des agents opposés à l’AS, agissent sur le gène SMN2 pour qu’il produise plus de protéine SMN, mais encore là, le nombre de copies de ce gène, un facteur déterminant de la gravité de cette maladie, joue un rôle important dans la réponse au traitement. En revanche, l’OA, qui fournit un gène SMN1 fonctionnel, s’attaque à la cause fondamentale de la maladie.
Administration d’un gène SMN1 fonctionnel en une seule injection
« L’OA est un agent de thérapie génique qui s’administre en une seule injection intraveineuse et qui fournit un gène SMN1 fonctionnel afin de rétablir l’expression d’une protéine SMN complète », a expliqué le Dr Richard D. Shell, pneumologue pédiatrique attaché au Nationwide Children’s Hospital, de Columbus, en Ohio. Lors du congrès de 2022 de la WMS, il a présenté de nouvelles données sur la faculté que possède l’OA de préserver le fonctionnement bulbaire, qui met en jeu des processus physiologiques élémentaires et néanmoins vitaux, dont la déglutition, la mastication et l’aptitude à protéger les voies respiratoires contre l’aspiration.
« [La thérapie génique] s’administre en une seule injection intraveineuse; elle fournit un gène SMN1 fonctionnel afin de rétablir l’expression d’une protéine SMN complète. »
Lors des études de phase III, les étapes du développement moteur (p. ex. s’asseoir sans aide, se tenir debout et marcher) des enfants dont l’AS avait été traitée pendant leurs premiers mois de vie se sont déroulées à un rythme typique chez les enfants. On peut donc légitimement s’attendre à ce que le fonctionnement bulbaire soit assuré quand le traitement est administré tôt, mais cette question n’a pas encore été évaluée en détail avec l’OA jusqu’à maintenant.
Analyse du fonctionnement bulbaire après la thérapie génique
Les analyses a posteriori des diverses études sur l’OA ont porté sur le fonctionnement bulbaire de 29 enfants traités tôt, avant l’apparition des symptômes, et sur celui de 65 enfants ayant reçu de l’OA seulement une fois les symptômes apparus. Chez ces derniers, les fonctions assurées par le bulbe rachidien n’ont pas toutes été consignées par les cliniciens-chercheurs, mais l’analyse a porté sur toutes celles qui l’ont été.
Dans les analyses a posteriori des données tirées de l’étude de phase III SPR1NT, il était entendu qu’un « fonctionnement bulbaire opérant repose sur l’intégrité des nerfs crâniens, celle-ci permettant à un individu d’acquérir des aptitudes pour la communication verbale et de satisfaire ses besoins nutritionnels tout en protégeant ses voies respiratoires », a expliqué le Dr Shell.
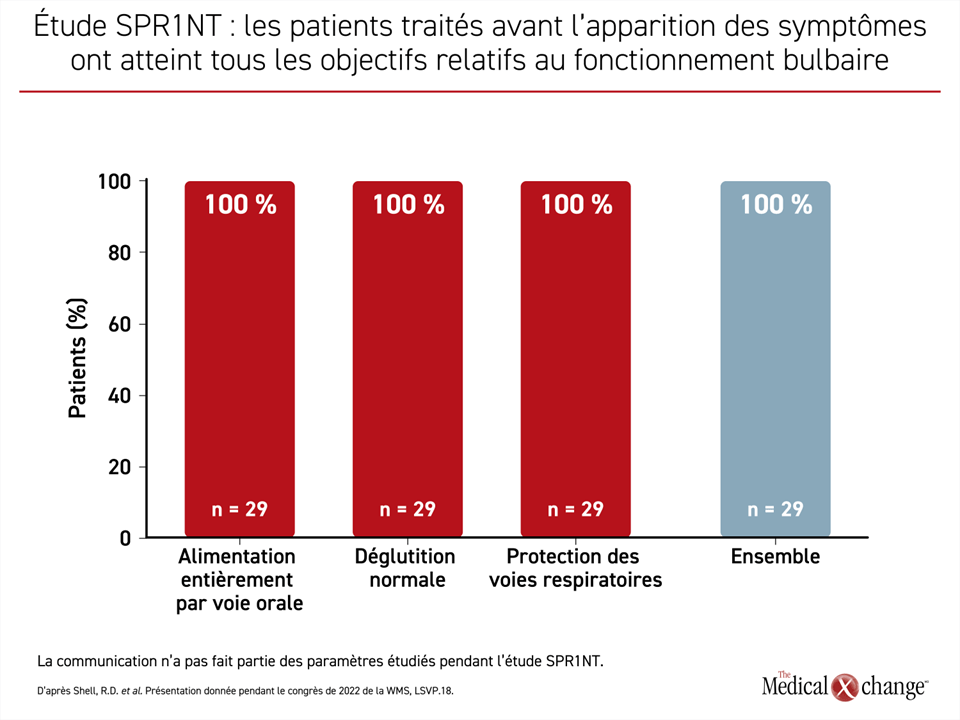
« Administré avant l’apparition des symptômes, l’OA a permis une alimentation entièrement orale, une déglutition normale et une protection totale des voies respiratoires. À preuve, aucun (0 %) des patients n’a subi aucun événement d’aspiration », a affirmé le Dr Shell (Figure 1).
« Administré avant l’apparition des symptômes, l’OA a permis une alimentation entièrement orale, une déglutition normale et une protection totale des voies respiratoires. À preuve, aucun (0 %) des patients n’a subi aucun événement d’aspiration »
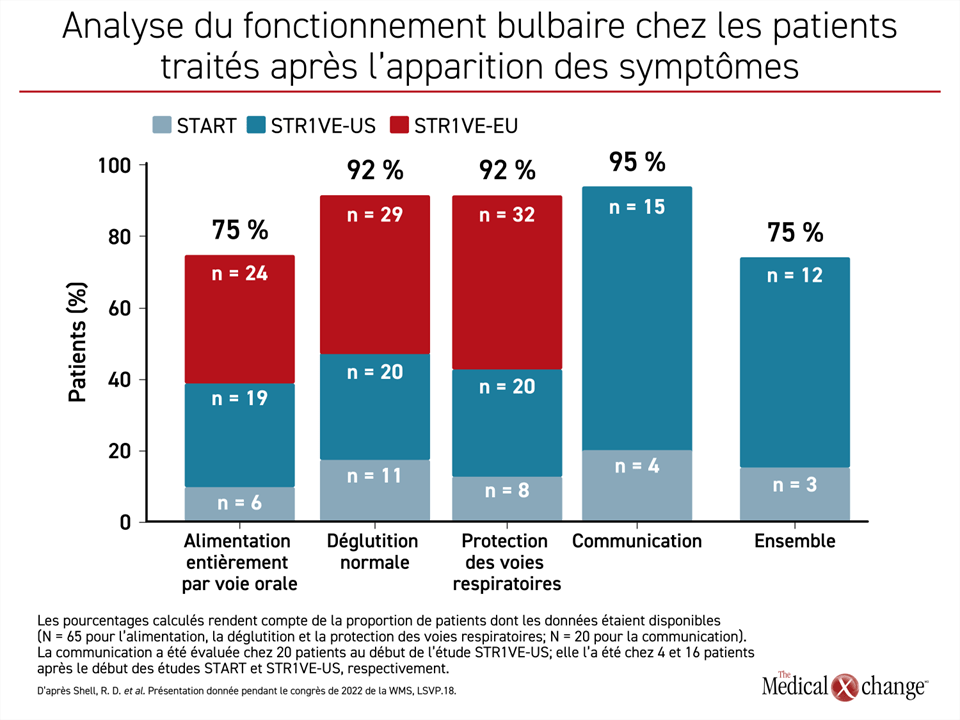
Quand l’OA a été administré après l’apparition des symptômes, comme ce fut le cas lors d’une étude de phase I (START) et de deux études de phase III (STR1VE-US et STR1VE-EU), les fonctions régies par le bulbe rachidien sont devenues opérantes chez beaucoup de patients, mais pas tous. En fait, 92 % des enfants déglutissaient et protégeaient leurs voies respiratoires normalement et pour de bon, alors que 75 % des enfants pouvaient se nourrir entièrement par voie orale. Il convient de noter que la communication verbale, qui n’est pas une fonction assurée par le bulbe rachidien, mais y est indirectement liée, était normale chez 95 % des patients (Figure 2).
Le traitement a des effets positifs plus marqués sur le fonctionnement bulbaire s’il est administré tôt
Selon le Dr Shell, ces observations confirment que l’idéal, pour le fonctionnement bulbaire et probablement aussi pour l’issue du traitement en général, consiste à administrer ce dernier avant l’apparition des symptômes.
Cela dit, les bienfaits exercés sur le fonctionnement bulbaire des enfants ayant reçu de l’OA après l’apparition des symptômes sont tout de même substantiels, selon la Dre Katlyn E. McGrattan, du Département de pédiatrie, de l’Université d’État de l’Ohio, à Columbus, en Ohio, qui a donné pendant le congrès de la WMS une présentation consacrée aux enfants traités par l’OA après l’apparition de leurs symptômes.
Dans ce groupe, certaines des fonctions assurées par le bulbe rachidien se sont émoussées au fil du temps : la protection des voies respiratoires est passée de 100 à 92 % et l’alimentation entièrement orale, de 85 à 75 % entre le début et la fin de l’étude. En revanche, les autres fonctions ont été préservées et en fait, se sont améliorées avec le temps. Par exemple, la capacité de communiquer est passée de 20 % à 95 % entre le début et la fin de l’étude.
« En intervenant tôt avec l’OA et une prise en charge interdisciplinaire, il semble possible de préserver et de stabiliser le fonctionnement bulbaire et les fonctions motrices même chez les patients aux prises avec une AS symptomatique », a déclaré la Dre McGrattan, qui a mené en collaboration avec le Dr Shell les deux études ayant servi à évaluer le fonctionnement bulbaire après l’administration d’OA. Selon elle, l’OA change le cours de l’AS.
« En intervenant tôt avec l’OA et une prise en charge interdisciplinaire, il semble possible de préserver et de stabiliser le fonctionnement bulbaire et les fonctions motrices même chez les patients aux prises avec une AS symptomatique »
« Les soins prodigués évoluent d’une démarche auparavant palliative à une démarche réadaptative », a ajouté la Dre McGrattan, soulignant au passage que plus le traitement est administré tôt, meilleurs sont les résultats.
Elle a toutefois tenu à préciser que la possibilité de rétablir la part du fonctionnement bulbaire qui a été perdue avec la thérapie génique semble restreinte. Il en serait de même pour les étapes du développement moteur telles que réussir à s’asseoir et à se tenir debout sans aide. Si ces étapes ne sont pas franchies à un âge normal, la fenêtre temporelle où ces aptitudes s’acquièrent semble se fermer avec peu de chances de réaliser des gains par la suite.
Selon les données du registre RESTORE, il faut intervenir tôt pour obtenir une efficacité et une innocuité optimales
Un registre mondial appelé RESTORE, qui est à la base une étude prospective d’observation menée chez des patients atteints d’AS et recevant un traitement de fond, indépendamment de la gravité de leur maladie, vient appuyer les données tirées des études. La plupart des 500 enfants et plus qui y sont inscrits jusqu’à présent possèdent 2 ou 3 copies du gène SMN2, une caractéristique épidémiologique de l’AS et signe d’une forme grave de la maladie et d’un pronostic sombre si elle n’est pas traitée.
Présentant une analyse des données du registre RESTORE lors du congrès de 2022 de la WMS, le Dr Laurent Servais, du Centre de médecine neuromusculaire MDUK Oxford, de l’Université d’Oxford, au R.-U., a déclaré : « Nous voulions établir l’efficacité et l’innocuité de l’OA utilisé en pratique clinique chez des enfants atteints d’AS qui l’ont reçu en monothérapie ou en remplacement du nusinersen ou d’un autre traitement de fond ».
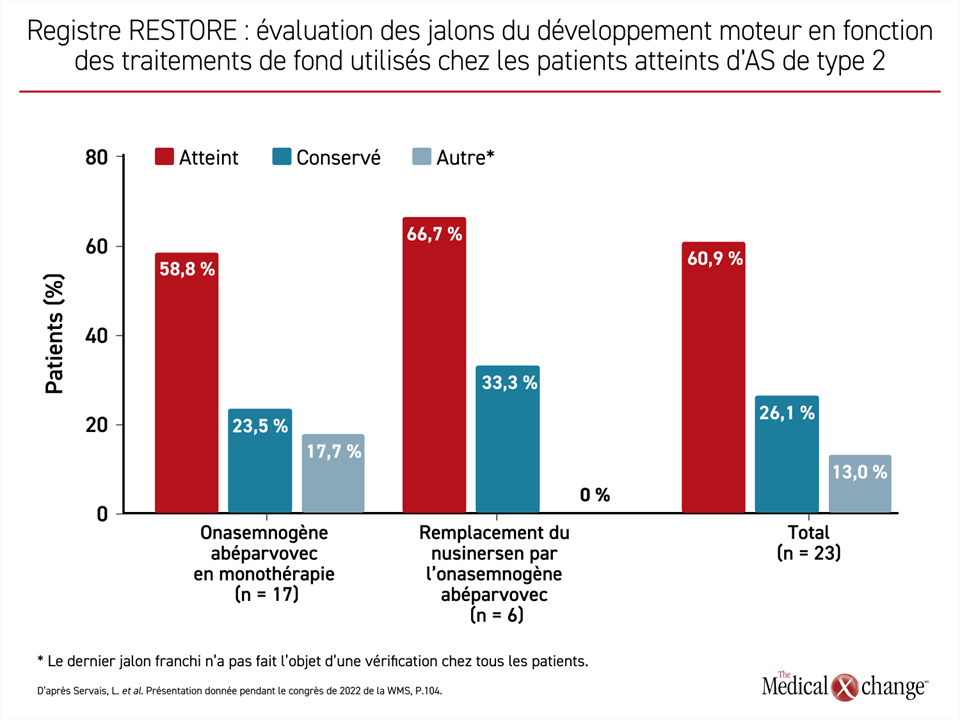
Parlant des 96 patients évaluables traités par de l’OA, le Dr Servais a affirmé que 89 d’entre eux ont franchi toutes les étapes de leur développement moteur après avoir été traités par cet agent et n’ont pas régressé jusqu’à maintenant. Sur ces 96 patients, 40 avaient reçu le traitement au long cours à base de nusinersen avant d’être traités par de l’OA. Ce traitement antérieur n’a pas eu d’influence majeure sur la réponse à l’OA (Figure 3). Au moment où certains de ces patients ont été traités, le nusinersen et l’OA étaient les seuls traitements de fond offerts contre l’AS. Il est rassurant de constater que le nusinersen n’atténue pas la réponse à l’OA qui peut ainsi être utilisé pour maîtriser l’AS durablement sans avoir à en administrer plusieurs doses.
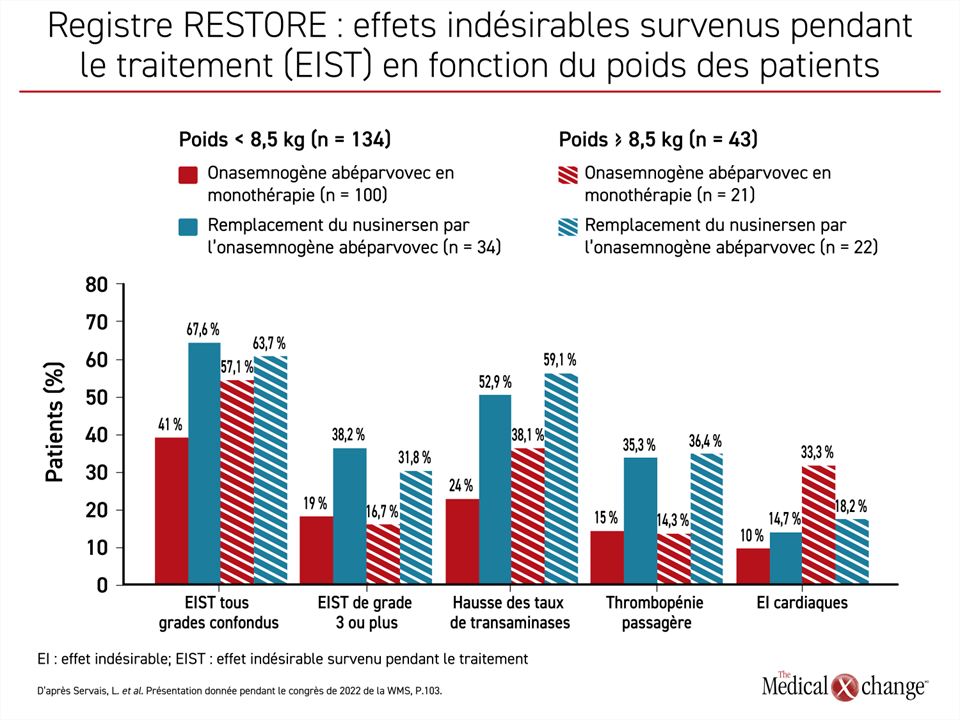
Selon le Dr Servais, l’emploi de l’OA en première intention plutôt qu’en remplacement d’un autre traitement de fond semble toutefois plus avantageux sur le plan de l’innocuité. La comparaison des effets indésirables observés chez les patients traités d’emblée par de l’OA et chez ceux ayant reçu cet agent en remplacement du nusinersen a montré que les effets indésirables de tous grades, ceux de grades 3 ou plus et ceux déjà reliés à l’OA, comme une hausse des taux d’enzymes hépatiques et une thrombopénie passagère, étaient moins répandus chez les premiers que chez les seconds (Figure 4).
La thérapie génique est efficace dans tout le spectre de l’AS
Deux autres séries de données tirées du registre RESTORE à l’appui de l’utilité de l’OA en pratique clinique ont été présentées dans le cadre du congrès de 2022 de la WMS. L’une d’elles montre que le poids des patients au moment où ils reçoivent l’OA n’a pas d’influence sur leur réponse à ce traitement, alors que l’autre a fait ressortir des gains chez les enfants atteints d’AS porteurs de 4 copies ou plus du gène SMN2.
Les enfants pesant au moins 8,5 kg ont été comparés à d’autres, de poids inférieur, aux fins d’évaluation de l’efficacité et de l’innocuité du traitement en fonction du poids des patients. Les enfants plus lourds étaient plus âgés et plus susceptibles d’avoir déjà subi un traitement de fond, ce qui explique probablement pourquoi la préservation des acquis en matière de développement moteur était plus marquée (45,8 % vs 19,1 %) que le franchissement des étapes de ce développement (45,9 % vs 74,6 %) dans ce groupe que dans celui des enfants qui l’étaient moins. Cela dit, l’amélioration objectivée à la fin de l’étude au moyen des scores CHOP-INTEND était considérable dans les deux groupes.
Les effets indésirables tous grades confondus étaient plus fréquents chez les enfants plus lourds (60,5 % vs 47,8 %), mais les effets indésirables graves l’étaient moins (18,6 % vs 21,6 %). D’après le Dr Servais, ces données doivent être interprétées avec prudence parce que les caractéristiques des patients n’étaient pas comparables et que la différence observée pour les effets indésirables de grade 3 ou plus et les effets indésirables graves n’était pas significative.
La deuxième série de données faisait état de l’efficacité et de l’innocuité de l’OA chez des patients atteints d’AS et porteurs d’au moins 4 copies du gène SMN2, signe d’une maladie relativement légère. Notons que les données de 14 patients seulement (9 étaient porteurs de 4 copies et 5 en possédaient au moins 4) ont pu être analysées. Il n’empêche qu’au dire du Dr Richard S. Finkel, du Centre des neurothérapies expérimentales, à l’Hôpital de recherche pédiatrique St-Jude de Memphis, au Tennessee, l’état de tous ces enfants s’est amélioré, peu importe la méthode d’évaluation utilisée, telle que l’échelle CHOP-INTEND, le franchissement de nouvelles étapes du développement moteur ou l’examen HINE-2 (Hammersmith Infant Neurological Examination-Part 2).
Quant à l’interprétation clinique de ces données, le Dr Finkel invite à la prudence, car selon lui la détermination du nombre de copies du gène SMN2 est peu fiable. Compte tenu des effets positifs de l’OA chez ces patients, il insiste sur le diagnostic précoce même chez les enfants porteurs d’un plus grand nombre de copies de ce gène.
« Ces données soulignent à quel point il est important de reconnaître cette maladie tôt et d’intervenir rapidement pour obtenir des résultats optimaux chez tous les patients atteints d’AS », a déclaré le Dr Finkel.
Conclusion
Les traitements de fond offerts depuis relativement peu de temps sauvent la vie des enfants atteints des formes graves d’AS, soit la majorité des cas de cette maladie. L’OA, le premier et le seul traitement de fond servant à remplacer le gène SMN1 dysfonctionnel, a jusqu’à maintenant permis de prévenir l’expression des symptômes de l’AS lorsqu’elle est traitée tôt dans la vie des enfants, d’où la préservation du fonctionnement bulbaire et le franchissement des étapes du développement moteur à un âge normal. Les données recueillies pendant des études et dans un registre témoignant de la pratique clinique montrent que s’il est administré après l’apparition des symptômes, même aux enfants ayant déjà reçu un autre traitement de fond, l’OA peut quand même agir sur le cours de la maladie.