Neurology
27th International Hybrid Annual Congress of the World Muscle Society (WMS)
Gene Replacement Therapy Fosters Age-appropriate Development in Spinal Muscular Atrophy
Halifax – Two sets of data show that a gene replacement therapy approved for the treatment of spinal muscular atrophy (SMA) permits age-appropriate development in most children if introduced in advance of symptoms. However, even when introduced after symptoms develop, substantial attenuation of progression is often achieved. These findings were initially reported in phase 3 trials that led to the regulatory approval of a gene replacement therapy, but registry data are showing the same benefits in a real-world setting. Follow-up accruing in both sets of data suggest that milestones once achieved are maintained long-term and perhaps indefinitely.
Prior to the relatively recent introduction of treatment, the most severe forms of SMA were uniformly fatal, usually before the age of 2 years. With treatment, children with SMA are living longer, but the gene replacement therapy onasemnogene abeparvovec (OA) has so far permitted age-appropriate development in many children, and not just survival. While OA is approved for treatment in children ≤21 kg based on the benefits observed in the registration trials, the phase 3 studies included some children enrolled within weeks of birth. In these children, most have achieved age-appropriate development milestones in follow-up so far, which in some cases extends several years.
For an exclusive interview with Dr. Laurent Servais on the impact to clinical practice, click here
SMA is characterized by inadequate levels of the SMN protein, which occurs because of dysfunction of the SMN1 gene. The SMN protein is essential to motor neurons that control bulbar function and many other motor functions. While the SMN2 gene can produce compensatory SMN protein, it is not sufficient. The available therapies for SMA, nusinersen and risdiplam, act on the SMN2 gene to increase SMN protein production, but, again, the number of SMN2 copies, which is a determinant of SMA severity, is important for response. In contrast, the delivery of a functional SMN1 gene by OA addresses the fundamental cause of disease.
Single-dose administration of a functional SMN1 gene
“OA is a one-time intravenously administered gene replacement therapy that delivers a functional SMN1 gene to restore expression of the full-length SMN protein,” explained Dr. Richard D. Shell, a pediatric pulmonologist affiliated with Nationwide Children’s Hospital, Columbus, Ohio. At the 2022 WMS meeting, he presented new data on the ability of OA to preserve bulbar function, which involves basic physiologic processes essential for life, including swallowing, chewing, and the ability to protect the airway from aspiration.
“[Gene replacement therapy] is a one-time intravenously administered [treatment] that delivers a functional SMN1 gene to restore expression of the full-length SMN protein.”
In the phase 3 trials, children with SMA treated within the first months of life have achieved motor milestones consistent with typically developing children, like sitting without assistance, standing, and walking at expected ages, so it is reasonable to expect that bulbar function is also achieved with early treatment. However, until now, this has not been examined in detail with OA.
Bulbar Function Analyzed After Gene Replacement Therapy
In post-hoc analyses from separate OA trials, bulbar function was evaluated in 29 children treated early in life and prior to the onset of symptoms as well as in 65 children who did not receive OA until they had already developed symptoms. In this latter group, not all bulbar functions were recorded by the study investigators, but the analysis included those that were.
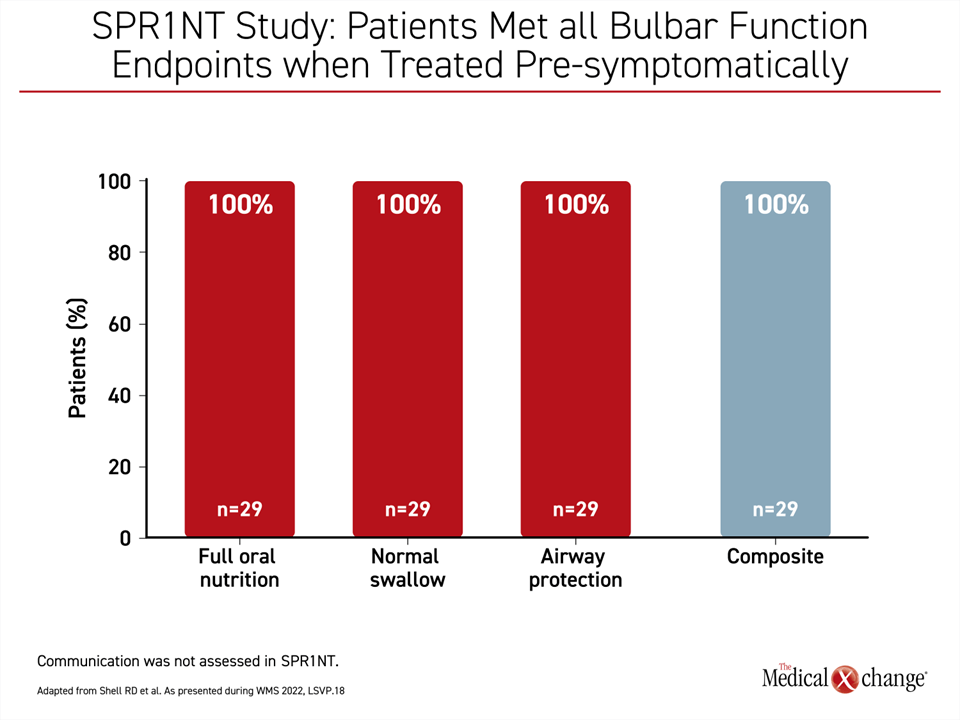
In post-hoc analyses of data from the Phase III SPR1NT study, “bulbar function was defined as integrity within cranial nerves that enables an individual to establish verbal communication skills and meet nutritional needs while maintaining airway protection,” Dr. Shell explained.
If administered pre-symptomatically, OA led to full oral nutrition, normal swallowing, and complete airway protection, meaning no aspiration events in 100% of patients,” Dr. Shell said (Figure 1).
“If administered pre-symptomatically, [gene replacement therapy] led to full oral nutrition, normal swallowing, and complete airway protection, meaning no aspiration events in 100% of patients.”
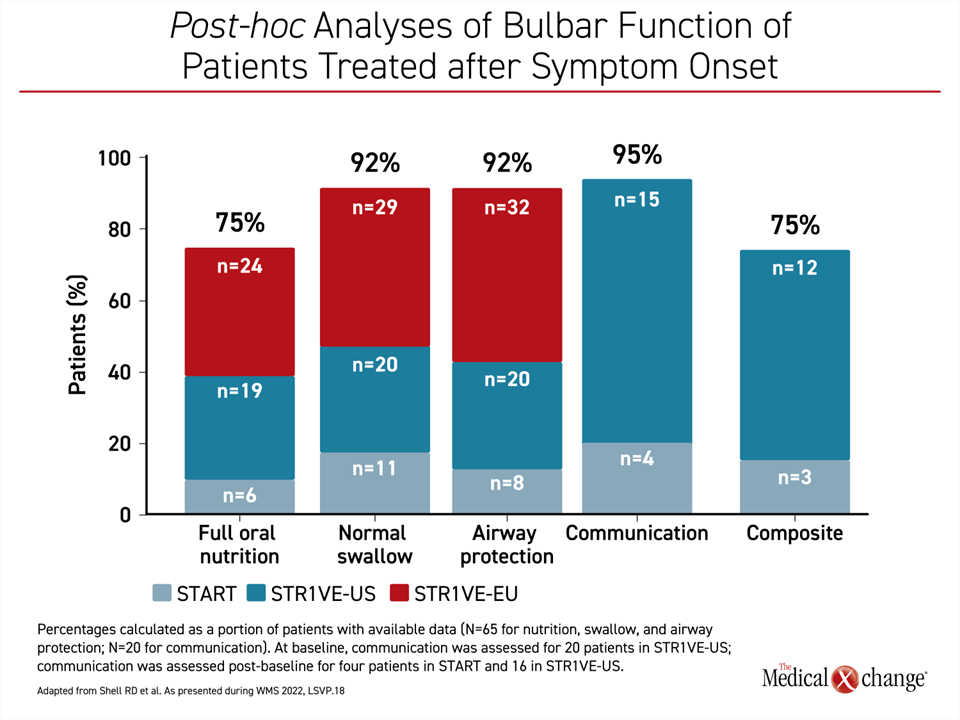
If administered after symptoms developed, as was the case in a phase I trial (START) and two phase 3 trials (STR1VE-US and STR1VE-EU), bulbar functions were achieved at substantial rates but not in all patients. Normal swallowing and airway protection was achieved and maintained by 92% of children, while 75% achieved full oral nutrition. It is notable that verbal communication, which is not a bulbar function but is tangentially related, was achieved in 95% of patients (Figure 2).
Increased Benefits in Bulbar Function with Early Therapy
Overall, these findings confirm that treatment before symptom onset was best from the perspective of bulbar function and presumably for outcome in general, according to Dr. Shell.
However, the benefits on bulbar function in children who did not receive OA until after symptoms developed were still substantial, according to Dr. Katlyn E. McGrattan, Department of Pediatrics, Ohio State University, Columbus, Ohio. She led a separate WMS presentation focused only on the children treated with OA after the onset of SMA symptoms.
In this group, there was a decline in some bulbar functions over time—airway protection declined from 100% to 92% and full oral nutrition declined from 85% to 75% from baseline to end of study—but other functions were protected and, in fact, improved over time. For example, ability to communicate increased from 20% at baseline to 95% at end of study.
“Early intervention with OA along with interdisciplinary management appears to preserve and stabilize bulbar and motor functions even in patients with symptomatic SMA,” reported Dr. McGrattan, who was a co-investigator with Dr. Shell on both studies evaluating bulbar function following OA. Overall, Dr. McGrattan said that OA is changing the trajectory of SMA.
“Early intervention with [gene replacement therapy] along with interdisciplinary management appears to preserve and stabilize bulbar and motor functions even in patients with symptomatic SMA.”
“Care is evolving from a palliative approach to a rehabilitative approach,” said Dr. McGrattan, who pointed out that the earlier the treatment, the better the outcome.
However, she cautioned that the ability of gene replacement therapy to reverse bulbar function that is already lost appears to be limited. The same has been observed of motor milestones, such as independent sitting and standing. If these milestones are not reached within the range of age-appropriate development, the window of acquisition appears to close with a low likelihood of subsequent gain.
RESTORE Registry Supports Early Intervention for Optimal Safety and Efficacy
A global registry called RESTORE, which is an ongoing prospective treatment-neutral study enrolling patients with SMA and following them in context with disease-modifying therapies (DMTs) regardless of disease severity, supports trial data. Consistent with the epidemiology of SMA, most children in the registry have just 2 or 3 copies of the SMN2 gene, meaning they have severe disease and a poor prognosis without treatment. There are more than 500 patients enrolled so far.
In a RESTORE analysis presented at the 2022 WMS, “we sought to describe the safety and effectiveness of OA in a real-world setting for children with SMA who received it as a monotherapy or after a switch from nusinersen or another DMT,” reported Dr. Laurent Servais, MDUK Oxford Neuromuscular Center, University of Oxford, UK.
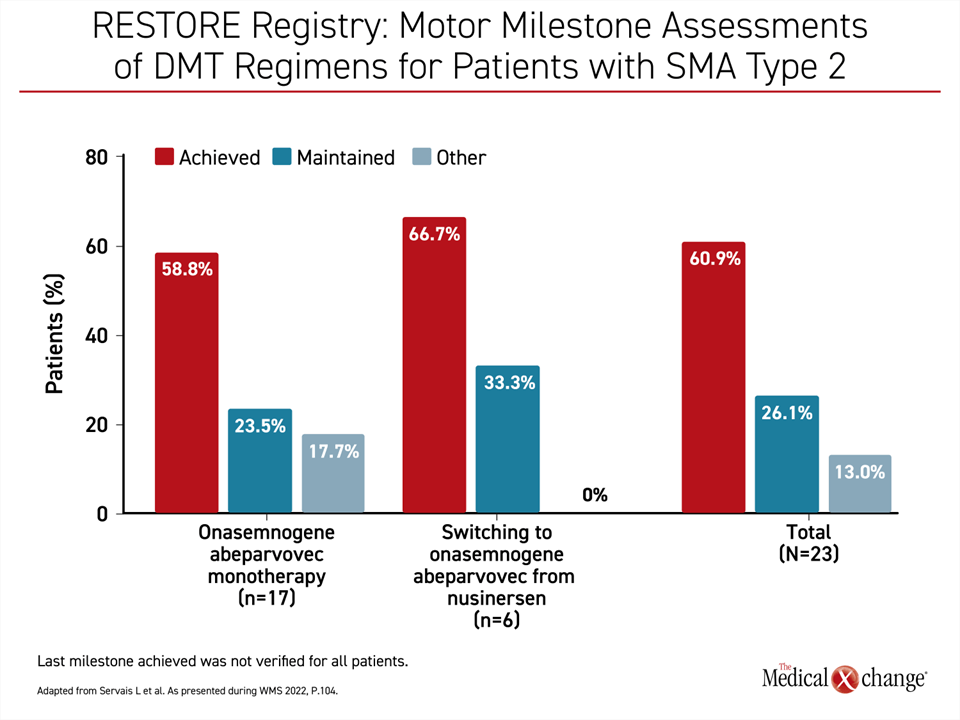
Presenting data on 96 evaluable OA patients, Dr. Servais reported that 89 have achieved or maintained all motor milestones once OA was started and through follow-up so far. Of these 96 patients, 40 had received the chronic therapy nusinersen prior to OA. Prior nusinersen treatment did not substantially influence the response to OA (Figure 3). At the time some patients were treated, nusinersen and OA were the only DMTs available for SMA therapy. The demonstration that it does not impact response to OA, which can still be employed to achieve long-term SMA control without repeated doses, is reassuring.
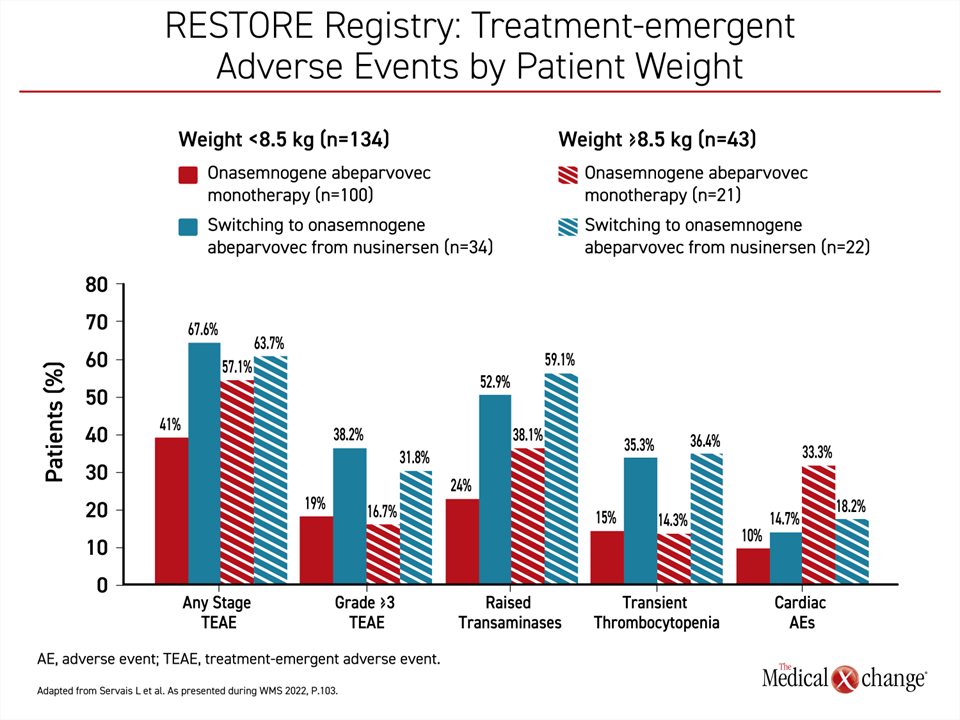
However, there appears to be a safety advantage for using OA as a first-line therapy rather than switching to OA after another DMT, according to Dr. Servais. When adverse events were compared among those who received OA as a first-line therapy and those who received OA following nusinersen, there was a lower rate of any stage adverse events, grade ≥3 adverse events, and adverse events previously associated with OA, including elevated liver enzymes and transient thrombocytopenia (Figure 4).
Gene Replacement Therapy Effective in Spectrum of SMA
Two other sets of data drawn from the RESTORE registry were presented at the 2022 WMS meeting. Both expand data on the value of OA in the real world. One showed that patient weight at the time of OA initiation does not influence response. The other showed benefits in children with SMA with ≥4 copies of the SMN2 gene.
In the efficacy and safety data based on weight, children weighing ≥8.5 kg were compared to those weighing less. The heavier children were older and more likely to have been treated with a prior DMT. This is likely to explain why the maintenance of motor milestones was more pronounced (45.8% vs. 19.1%) than the acquisition of milestones (45.9% vs. 74.6%) when heavier children were compared to lower-weight children. However, the end-of-study improvement in CHOP-INTEND scores were substantial in both groups.
Adverse events of any grade were more common in heavier children (60.5% vs. 47.8%) but serious adverse events were lower (18.6 vs. 21.6%). Dr. Servais recommended a cautious interpretation of these adverse event data because patient characteristics were not balanced and because there was no significant difference observed for grade 3 or higher or serious adverse events.
In a second set of data, OA efficacy and safety data were analyzed in SMA patients with ≥4 copies of the SMN2 gene, which signals relatively mild disease. The numbers were small. Only 14 patients (nine with 4 copies, and five with ≥4 copies) were available for analysis, but all had improvement with various methods of evaluation, such as CHOP-INTEND, acquisition of new motor milestones, or the Hammersmith Infant Neurological Examination-Part 2 (HINE-2), according to Dr. Richard S. Finkel, Center for Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, Tennessee.
For a clinical interpretation of these data, Dr. Finkel cautioned that the determination of SMN2 gene copies is unreliable. Given the benefit with OA in these patients, he suggested that this keeps the focus on early diagnosis even among those with a greater number of SMN2 gene copies.
“These data highlight the importance of early identification and intervention to optimize outcomes for all SMA patients,” Dr. Finkel said.
Conclusion
The relatively recent introduction of DMTs for SMA are saving the lives of children with severe forms, which accounts for the majority of the SMA population. OA, the first and only DMT to replace the dysfunctional SMN1 gene, has so far prevented expression of symptoms when SMA is treated early in life. This includes preservation of bulbar function and achievement of motor milestones at expected ages. If initiated after symptoms, even in patients with prior experience with another DMT, OA can still modify the disease course, according to both trial and real-world registry data.