neurology
64th Annual Meeting of the American Academy of Neurology (AAN)
Challenge of Therapy Selection in Multiple Sclerosis Increases with Rapid Expansion in Drug Classes
New Orleans – New and emerging therapeutic options for multiple sclerosis (MS) will eventually demand a re-evaluation of first-line algorithms. For now, newer disease-modifying therapies (DMTs), including an oral drug, have been largely relegated to second- or third-line therapy until longer follow-up can increase evidence that they are well tolerated and safe when taken chronically. Vigilance for long-term safety is important for all medicines on which patients are likely to remain indefinitely, but DMTs in MS require particular caution due to their effect on immunomodulatory processes that mediate defenses against infection and malignancy. The eventual transition from established DMTs, which have proven protection against relapse with a low risk of serious adverse events, to newer DMTs will be dependent on confidence that newer drugs not only offer equal protection against MS progression with low risks of side effects but do not adversely alter innate immune defenses.
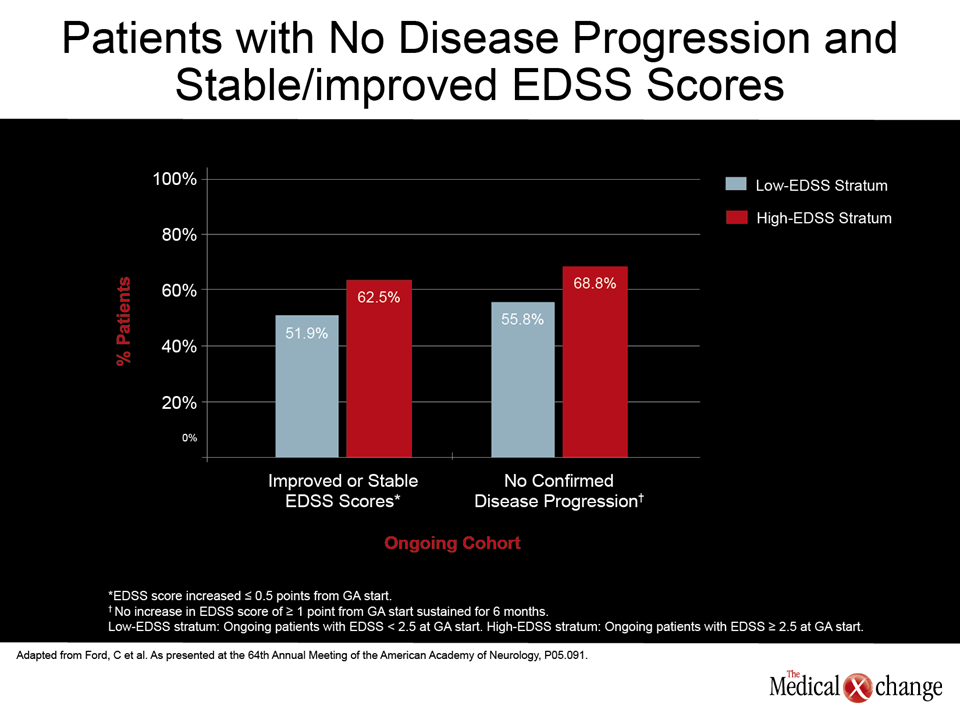
During this year’s American Academy of Neurology (AAN) meeting, data was presented on a variety of new and emerging therapies for multiple sclerosis (MS), including fingolimod, BG-12, teriflunomide, laquinimod, and alemtuzumab. These data were joined by additional studies attempting to establish where the first-line agents, glatiramer acetate (GA) and the interferons, fit within strategies to provide disease control within maximum tolerability and safety. A novel study suggesting that genetic profiling may be useful for predicting treatment response contribute to evidence that first-line management of MS may eventually evolve (Macciardi F et al. AAN 2012, Abstract S20.003). However, safety and tolerability remain the focus for current first-line treatment selection until there is better evidence that newer agents offer advantages without introducing new risks. “We do not fully understand all of the effects of any of the disease-modifying agents, but we do know that we are altering immune cell populations and inflammatory processes. The importance of long-term follow-up in confirming the safety of these agents is hard to underestimate. We really do not know what the consequences might be on various organ systems from altering immune function over very prolonged periods,” reported Dr. Corey Ford, Professor of Neurology, University of New Mexico, Albuquerque. The remarks from Dr. Ford were made in the context of a study he presented on very long-term follow-up in a patient population that started disease-modifying therapy (DMT) with an Expanded Disability Status Scale (EDSS) score of 2.5 or greater (Ford, C et al. AAN 2012, P05.091). These data, more difficult to collect today, were generated from a prospective study of GA that was initiated in 1991, when there were no approved DMTs. At the time, patients often had advanced MS at the start of therapy. In this latest analysis, 48 patients with an EDSS of at least 2.5 were compared to 52 patients with a lower EDSS at the start of therapy. The mean GA exposure was 13.6 years with a range of 11.7 to 15.3 years. After this length of follow-up, “two thirds of those with a high baseline EDSS score [mean 3.58] had no confirmed disability progression and three quarters remained ambulatory without a walking aid,” Dr. Ford reported. “Natural history studies would predict that those with a high baseline EDSS score would have an accelerated course to disability with about one half requiring a walking aid within 10 years. Instead, the progression in those with a high EDSS was similar to those with a low EDSS (Fig. 1).” The protection from disease progression even in patients with substantial disability at the start of therapy has important implications for prolonged disease control, but Dr. Ford also reported “no unexpected safety issues” in more than a decade of follow-up with the DMT used in this study.
Evaluating Long-Term Safety
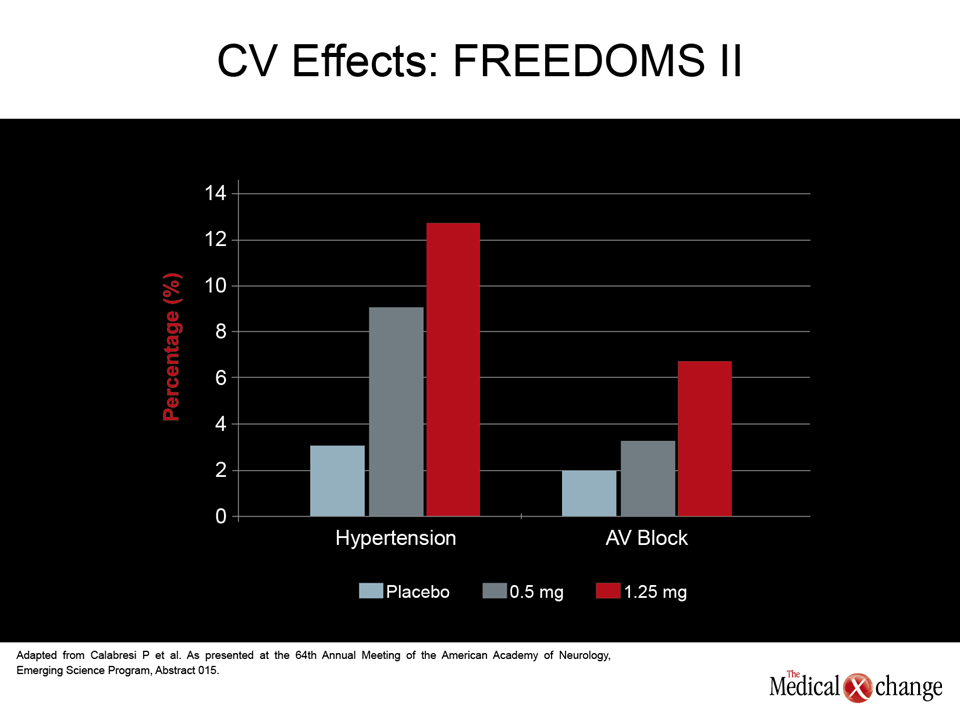
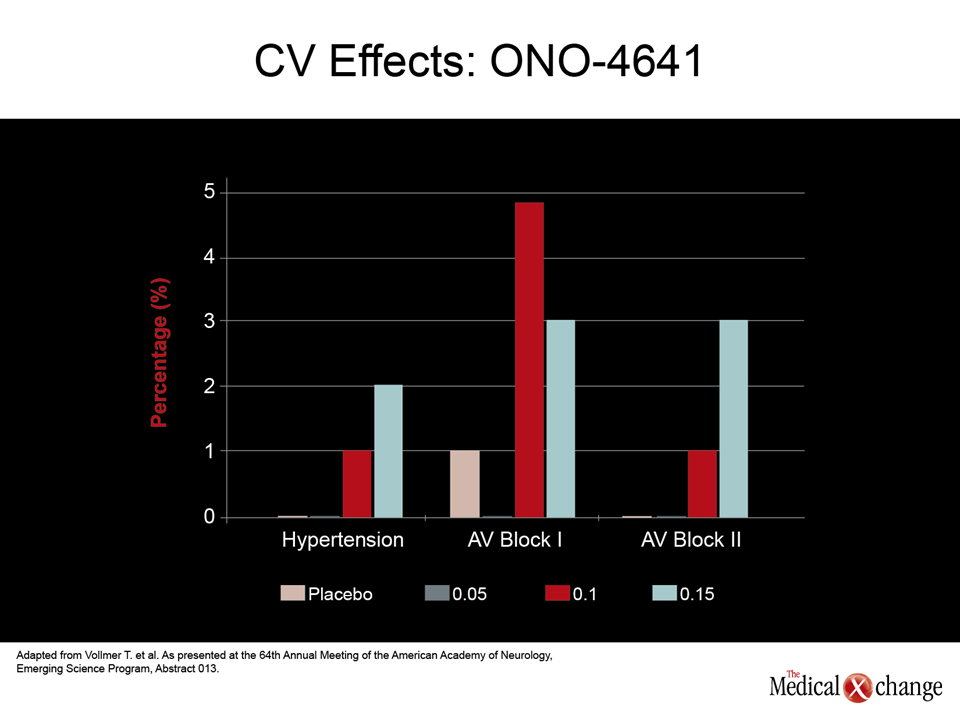
Of long-term safety issues, the immunomodulatory changes thought to underlie malignancy, serious infections, or progressive multifocal leukoencephalopathy (PML), are among the most worrisome, but the risk of significant adverse events not directly related to immune function may also limit the utility of MS drugs employed indefinitely. The relative importance of gastrointestinal (GI) and cardiovascular (CV) side effects among newer agents are being evaluated intensively. Recently, the U.S. Food and Drug Administration (FDA) strengthened the cardiac labeling for fingolimod, the first licensed oral DMT, based largely on changes in blood pressure and heart rate identified in phase III studies. Rather than monitoring at baseline and 6 hours after a first dose, the new labeling requires hourly measurements of blood pressure and heart rate over an initial 6-hour observation period. CV data from the FREEDOMS II study with the oral agent fingolimod presented at the 2012 AAN meeting reinforce this caution (Calabresi PA et al. AAN 2012, Emerging Science Session 015). According to results presented by Dr. Peter Calabresi, Director, Multiple Sclerosis Center, Johns Hopkins University, Baltimore, MD, the rates of hypertension for the placebo, 0.5 mg once-daily, and 1.25 mg once-daily arms were 3.1%, 8.9%, and 12.7%, respectively. The rates of atrioventricular (AV) block were 2%, 3.4%, and 6.7%, respectively (Fig. 2). In the FIRST study, which was specifically designed to track changes in blood pressure and heart rate with evaluations performed in 2289 patients, the rate of Mobitz type 1 heart block on fingolimod was 1.4%, according to Dr. Giancarlo Comi, Director, Department of Neurology, University Vita-Salute San Raffaele in Milan, Italy. While the rate of Mobitz type 2 heart block was <0.1%, there was a greater risk of bradycardia and adverse CV changes in those with heart disease at baseline (Comi et al. AAN 2012, S41.003). In the phase III FREEDOMS study, presented by Dr. Ludwig Kappos, Professor of Neurology, University of Basel, Switzerland, CV events in the fingolimod arm, including bradycardia and AV block, were observed but did not lead to any clinically significant consequences (Kappos et al. AAN 2012, S41.004). The cardiac effects of fingolimod, which is a selective S1P modulator, may or may not be class specific. For example, placebo-controlled phase II data presented here with ONO-4641, an experimental agent in this class, did not identify hypertension as a common side effect even at the highest dose (0.15 mg daily) relative to placebo (2% vs. 0%), but first-degree AV block did appear to occur more frequently on both the 0.15 mg dose (3%) and the 0.1 mg dose (4.9%) relative to placebo (1%), according to Dr. Timothy Vollmer, Professor of Neurology, University of Colorado, Denver (Fig. 3) (Vollmer TL et al. AAN 2012, 5LB001.013). In a phase II double-blind data generated with BAF312, another promising S1P modulator, bradycardia, along with dizziness and headache, were listed among the most common side effects in data presented by Dr. Olaf Stüve, Associate Professor of Neurology, Southwestern Medical Center, Dallas, TX (Stüve O et al. AAN 2012, S30.001) .
Tolerability
BG-12, another oral agent reaching advanced clinical testing, has a different mechanism of action and has not been associated with a substantial increase in cardiac risk. The major adverse effects of this agent, which is a fumaric acid ester, have been flushing, diarrhea, and nausea, according to the phase III CONFIRM data presented at the 2012 AAN by Dr. J. Theodore Phillips, Director, Multiple Sclerosis Center, Texas Neurology, Dallas, Texas (Phillips, JT et al. AAN 2012, S41.005). In this study, a BID 240 mg dose of BG-12, a TID 240 mg dose of BG-12, and GA were compared to placebo. The incidences of flushing were 31%, 24%, 4%, and 2% for BID BG-12, TID BG- 12, placebo, and GA, respectively. The incidences of diarrhea were 13%, 15%, 8%, and 4%, respectively, and the incidences of nausea were 11%, 15%, 8%, and 4%, respectively. No new safety data was presented at the 2012 AAN on teriflunomide, an oral DMT in development that inhibits pyrimidine synthesis by blocking dihydroorotate dehydrogenase, but a novel analysis of the previously completed Teriflunomide Multiple Sclerosis Oral (TEMSO) trial (O’Connor P. et al New Engl J Med 2011;365;1293-303) by Dr. Aaron Miller, Mount Sinai School of Medicine, New York City, NY, did find that teriflunomide reduced the annual rate of relapses leading to hospitalization by 43% (P<0.001) relative to placebo (Miller, A et al. AAN 2012, S30.003). In the original TEMSO data, which compared 7 mg teriflunomide and 14 mg teriflunomide to placebo, diarrhea, which occurred in 14.7% of those on the lower dose, 17.9% of those on the higher dose, and 8.9% of those on placebo, was a safety concern. Nausea was slightly more common on the higher dose of teriflunomide relative to placebo (13.7% vs. 7.2%). Hair thinning or decreased hair density occurred in 10.3% of those on the lower dose of teriflunomide, 13.1% of those on the higher dose, and 3.3% of the placebo group.
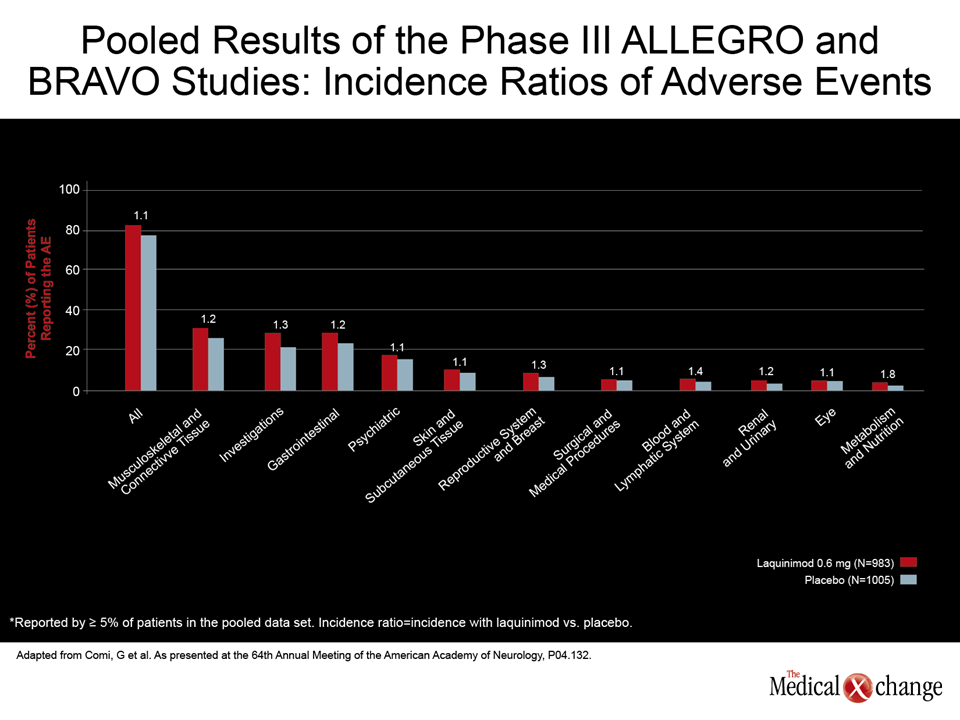
Pooled safety analyses from the Assessment of Oral Laquinimod in Preventing Progression in Multiple Sclerosis (ALLEGRO) and Benefit Risk Assessment of Avonex and Laquinimod (BRAVO) trials with laquinimod, another oral DMT in development, did not reveal any significant increase in CV or GI events (Comi, G et al. AAN 2012, S04.132). According to the pooled data, presented by Dr. Comi, 0.5% of patients randomized to placebo versus 0.3% of those randomized to laquinimod with normal electrocardiogram (ECG) at baseline developed ECG abnormalities. Liver enzyme elevations of greater than 3 times the upper limit of normal were more common on laquinimod than placebo (4.5 vs. 1.9%), but discontinuation rates for this adverse event was similar as was the proportion of patients with liver enzymes more than 5 times the upper limit of normal (1.1% vs. 1.3%). Dr. Comi, who also noted that the rates of serious adverse events were comparable on laquinimod relative to placebo, characterized the safety profile of this experimental oral agent as “excellent” (Fig. 4).
In phase III data from the Comparison of Alemtuzumab and Rebif Efficacy in Multiple Sclerosis (CARE- MS) I and II studies with alemtuzumab, an injectable monoclonal antibody that targets the CD52 antigen on T and B cells, appears to induce an autoimmune response that affects thyroid function. Again, this agent, like other newer DMTs in late stages of development, has demonstrated encouraging clinical efficacy, but the long-term safety may ultimately determine its place in a treatment algorithm. In CARE-MS II data presented here by Dr. Krzysztof Selmaj, Department of Neurology, Medical University of Lodz, Poland, thyroid-related autoimmune adverse events were 18.1% in the alemtuzumab group versus 6.4% on those randomized to subcutaneous interferon beta- 1a (SC IFNB-1a). Immune thrombocytopenia (ITP) was reported for 0.8% vs. 0.5%, respectively, when determined by platelet counts (Selmaj K et al. AAN 2012, Platform Presentation S41.006). Although alemtuzumab appeared to be more effective than IFNB1a in both CARE-MS I and II studies, the monitoring for autoimmunity over time will be pursued intensively.
While safety will be a critical variable in efforts to reconsider whether the order of first-line therapies should be altered with the availability of new agents, a genome-wide association study (GWAS) has generated intriguing evidence that therapies may be individualized to maximize efficacy in the context of safety. Conducted with GA, the GWAS identified 31 single nucleotide polymorphisms (SNPs) which were associated with a super response or super non-response to GA. From these, a 6 SNP model was developed to test patients for a super response to GA, defined as no relapse, no new T2 lesions, and no enhancing lesions at 12 months (Macciardi F et al. AAN 2012, Abstract S20.003). When tested in consenting patients from clinical trials with GA, the model had a 90.6% positive predictive value and a 90.5% negative predictive value for super responders, according to Dr. Fabio Macciardi, Chair, Genomics Working Group, University of California, Irvine. “These are important studies to pursue. At this point, they introduce more questions than they answer, because the variables that affect response in a single individual to a DMT are likely to be so complex, but individualized therapy is attractive, and the authors can be congratulated on pursuing this path,” commented Dr. Jack Antel, Professor, Montreal Neurological Institute, Montreal, QC.
Conclusion
The large increase in the number of therapies that are demonstrating activity against MS suggests that treatment algorithms may change as more is understood about the relative strengths and weaknesses of these agents. The cornerstone DMTs, GA and the interferons, are expected to continue to play an important role in the treatment of MS unless newer agents prove to be as effective, as well tolerated and equally safe.