Neurology
CURE SMA: Research and Clinical Care
Meeting 2021
Gene Therapy for Spinal Muscular Atrophy Effective as First- or Second-line Therapy
Virtual Meeting – A currently approved gene replacement therapy for spinal muscular atrophy (SMA) is demonstrating efficacy in children who have already received another SMA treatment as well as in children older than those who participated in the pivotal studies. Although the gene replacement therapy is approved for use up to 24 months of age, most participants in the initial and pivotal studies were treated within months of birth, most often before symptoms developed. These studies were also largely restricted to type 1 SMA, which is the most common and most severe form. The new data suggest benefit in a much broader range of patients.
Gene therapy for type 1 SMA has proven very effective. In a population with a life expectancy of less than two years, there are now children followed for more than five years with normal or near normal development. When it was approved, the gene replacement therapy onasemnogene abeparvovec, which replaces the SMN1 gene to restore SMA protein, became the second licensed treatment for a challenging disease. The first therapy was nusinersen, an antisense SMN2 splicing therapy that requires every four-month intrathecal maintenance dosing. Gene replacement therapy is administered only once.
RESPONSE TO GENE THERAPY IN OLDER CHILDREN
“Both therapies have dramatically improved the prognosis of SMA, but nearly all the patients in the clinical trials with onasemnogene abeparvovec were less than six months of age without any prior treatment,” reported Dr. Eric Wu, Researcher and Principal Investigator, Analysis Group, Boston, Massachusetts. Benefit in older children or in children previously exposed to nusinersen was not evaluated in these studies, but now, “due to real-world data in both of these groups of patients, we have some limited experience.”
“Due to real-world data, we now have some limited experience with gene therapy in older children and children previously treated with nusinersen.”
One set of data come from an interim analysis in 14 patients. All had been treated in children at least six months of age. Four of the patients received gene therapy as a first-line treatment. The other 10 were switched to onasemnogene abeparvovec after initial treatment with nusinersen. Most of the switchers (80%) had SMA type 1. In the onasemnogene abeparvovec monotherapy group, 25% had type 1, 25% had type 2, and 50% had type 3. Outcomes were evaluated by chart review. None of the patients had participated in a clinical trial or in the RESTORE registry, which is an on-going multinational observational study of onasemnogene abeparvovec with a planned follow-up of 15 years.
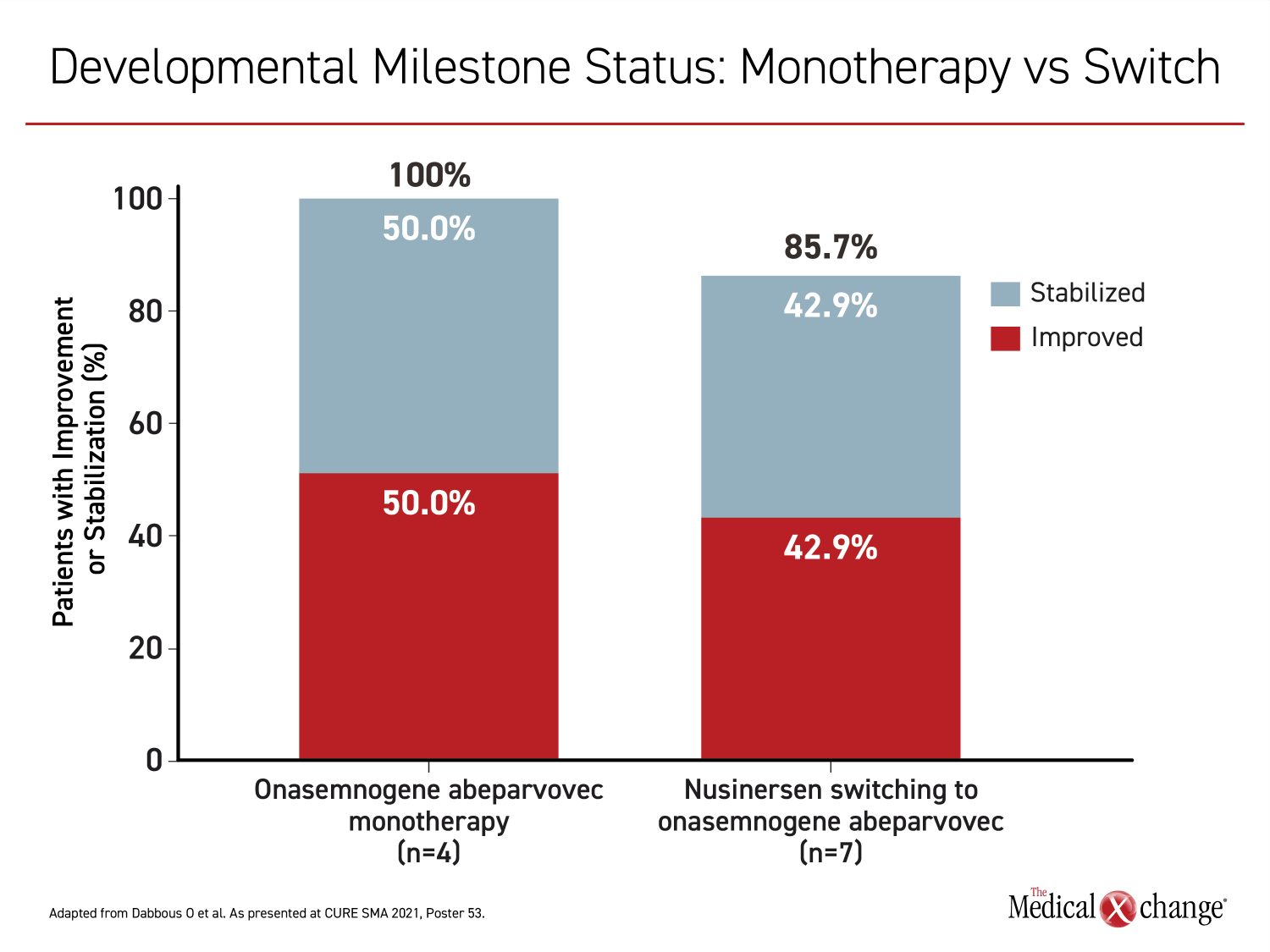
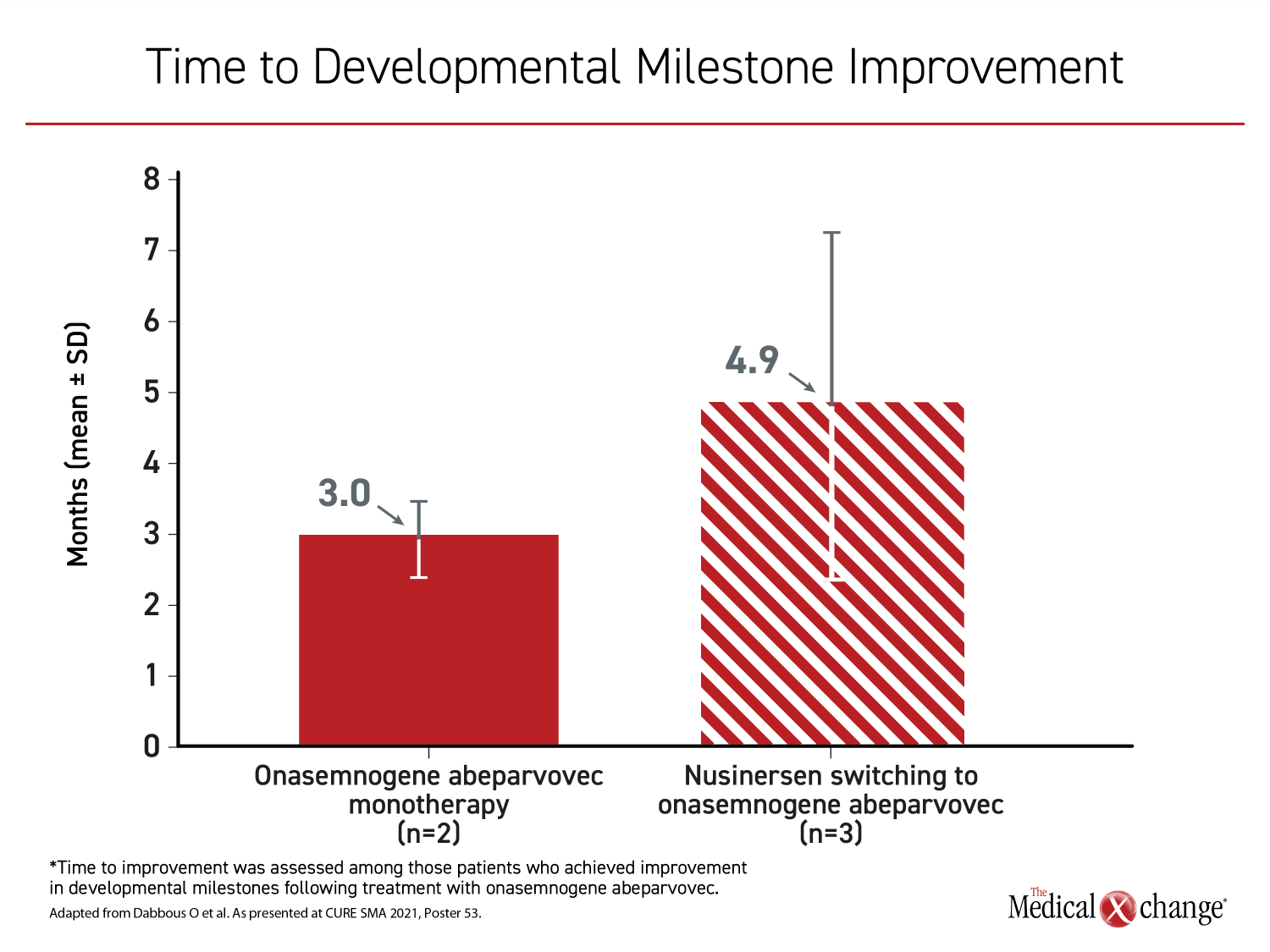
Similar in the two groups, the mean age at initiation of onasemnogene abeparvovec was slightly older than 17 months with a range of about 9 months to 24 months. Among those in the onasemnogene monotherapy group, developmental milestones were achieved in all four patients. In seven of 10 switchers for which developmental milestones were available, six (86%) achieved a new developmental milestone, such as eating without assistance or speaking, after the switch. The time to a new milestone on treatment was longer in those switched than in those receiving onasemnogene abeparvovec as a first-line monotherapy (3.0 vs. 4.9 months) (Figure 1) and (Figure 2).
IMPROVEMENT WITH FIRST- OR SECOND-LINE GENE THERAPY
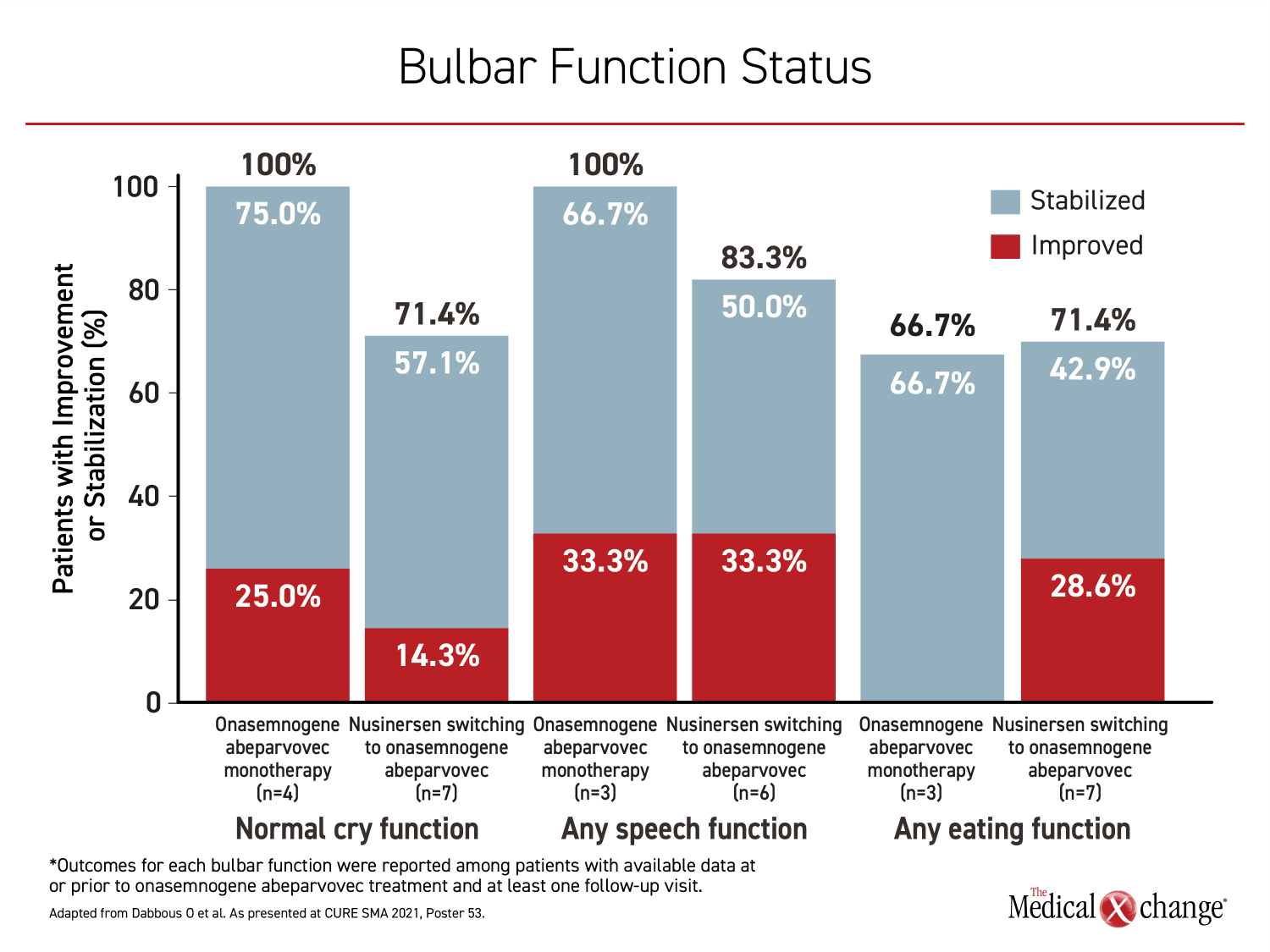
For specific functions evaluated in relation to therapy, most were improved or stabilized on gene replacement therapy whether it was used for first- or second-line. Following nusinersen, 33.3% of patients on gene replacement therapy improved speech function, while 50% stabilized. Furthermore, 14.3% improved cry function, while 57.1% stabilized and 28.6% improved eating function, while 42.9% stabilized. For those initiated on onasemnogene abeparvovec, the proportions with these functions at follow-up were 100%, 100%, and 66.7%, respectively (Figure 3).
REAL-WORLD DATA SHOW FAVOURABLE BENEFIT-TO-RISK PROFILE IN CHILDREN >6 MONTHS OF AGE
A second real-world analysis of gene replacement therapy in older SMA children also presented at the meeting evaluated 69 children in the RESTORE registry first treated after the age of 6 months. About half were treated between six and 12 months. Most of the remaining were treated between 12 and 24 months of age. The majority (65%) had SMA type 1. Thirty percent of the patients had three SMN2 copies, while almost all the others had two SMN2 copies.
Of these patients, 22 (31%) received onasemnogene abeparvovec as a first-line monotherapy. Of the remaining, 28 (40%) were switched to onasemnogene abeparvovec after first receiving nusinersen or risdiplam, a more recently developed oral SMN2 splicing therapy. Most of the others received SMN2 gene splicing therapy both before and after onasemnogene abeparvovec. Clinical responses were evaluated with the validated HINE-2 (Hammersmith Infant Neurological Examination) and CHOP INTEND (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders) tools for neurological and motor function.
96% PATIENT IMPROVEMENT ON CHOP INTEND SCORE
“Of 23 patients with two or more CHOP INTEND assessments, 22 (96%) improved or maintained their score over the study period,” reported Dr. Laurent Servais, Department of Pediatrics, Oxford Neuromuscular Center, Oxford, UK.
In a follow-up of up to two years, 19 (83%) of the patients had an increase in CHOP INTEND score, of which 15 (65% of the total patients) had a gain of at least four points. Only four patients had two or more assessments with HINE-2. One patient had a “marginal” response, but a gain in score was achieved for the other three, according to Dr. Servais.
In this real-world evaluation, rates of adverse events were similar when patients treated between six and 12 months of age were compared to those treated at an older age. Many adverse events, such as heart rate changes and elevated troponin, have been without adverse cardiac outcomes or other clinically meaningful events. All cases of hepatotoxicity, which typically involved elevations of liver enzymes but included acute liver failure, have resolved with prednisolone. A death due to respiratory arrest was not considered treatment related. Thrombocytopenia has been reported, but all cases have resolved without intervention, Dr. Servais reported.
“Based on the available data to date, the benefit-risk profile of onasemnogene abeparvovec remains favorable even in patients treated after six months of age,” Dr. Servais said.
“Based on the available data to date, the benefit-risk profile of onasemnogene abeparvovec remains favorable even in patients treated after six months of age.”
MILESTONES MAINTAINED IN LONG-TERM FOLLOW-UP
The data in older SMA patients and previously treated patients joins extended follow-up with those in the first group ever treated with onasemnogene abeparvovec, as well as those who participated in the clinical studies that led to regulatory approval. The former group, which is being followed in a cohort called LT-001, now has follow-up data beyond five years. The latter group is being followed in a study called LT-002. In both, the remarkable gains in developmental milestones on treatment have been sustained.
“During follow-up so far, no patient has lost a milestone that they had achieved previously,” according to Dr. John W. Day, Professor of Neurology, Stanford University Medical Center, California.
Evidence of benefit is typically observed within weeks to months after the gene therapy is administered. Although some gains are not observed immediately, Dr. Day reported even late responses, once gained, have not been lost. In one example of a late response in the LT-001 population, treatment had been initiated in a child who had already developed significant neuromuscular weakness. Improvement was slow, but the ability to stand with support was achieved approximately 42 months later. This milestone has not been lost in follow-up that now extends for an additional two years.
RESPONSE TO GENE THERAPY IS STABLE
“What has been seen so far is a stability of response, so there is a duration of effect that has been sustained for the period of time that we have studied them, which included durable control in children up to 6.1 years of age in the LT-001 study,” Dr. Day reported.
In LT-002, which includes the STRIVE and SPR1NT pivotal studies that were considered for regulatory approval of onasemnogene abeparvovec, follow-up now extends to 2.7 years. Reviewing the experience from both studies, Dr. Day reported that many patients are continuing to meet all age-appropriate milestones with “no fall off for any functional gains” so far. As a result, none of the patients who participated in the STRIVE or SPR1NT studies are now receiving any forms of ventilatory or feeding tube support in current follow-up.
By restoring the function of the SMN1 gene, onasemnogene abeparvovec addresses the underlying defect of SMA, restoring the deficient protein that leads to neuromuscular degeneration. While the SMN2 gene splicing therapies can also increase circulating levels of the SMN protein, the SMN2 gene is less efficient for SMN protein production than the SMN1 gene. Although unproven, gene therapy is potentially curative, while the SMN2 gene splicing therapies must be maintained indefinitely. This might be an important limitation in the context for scheduled dosing whether intrathecal or oral.
TREATMENT PERSISTENCE EXAMINED
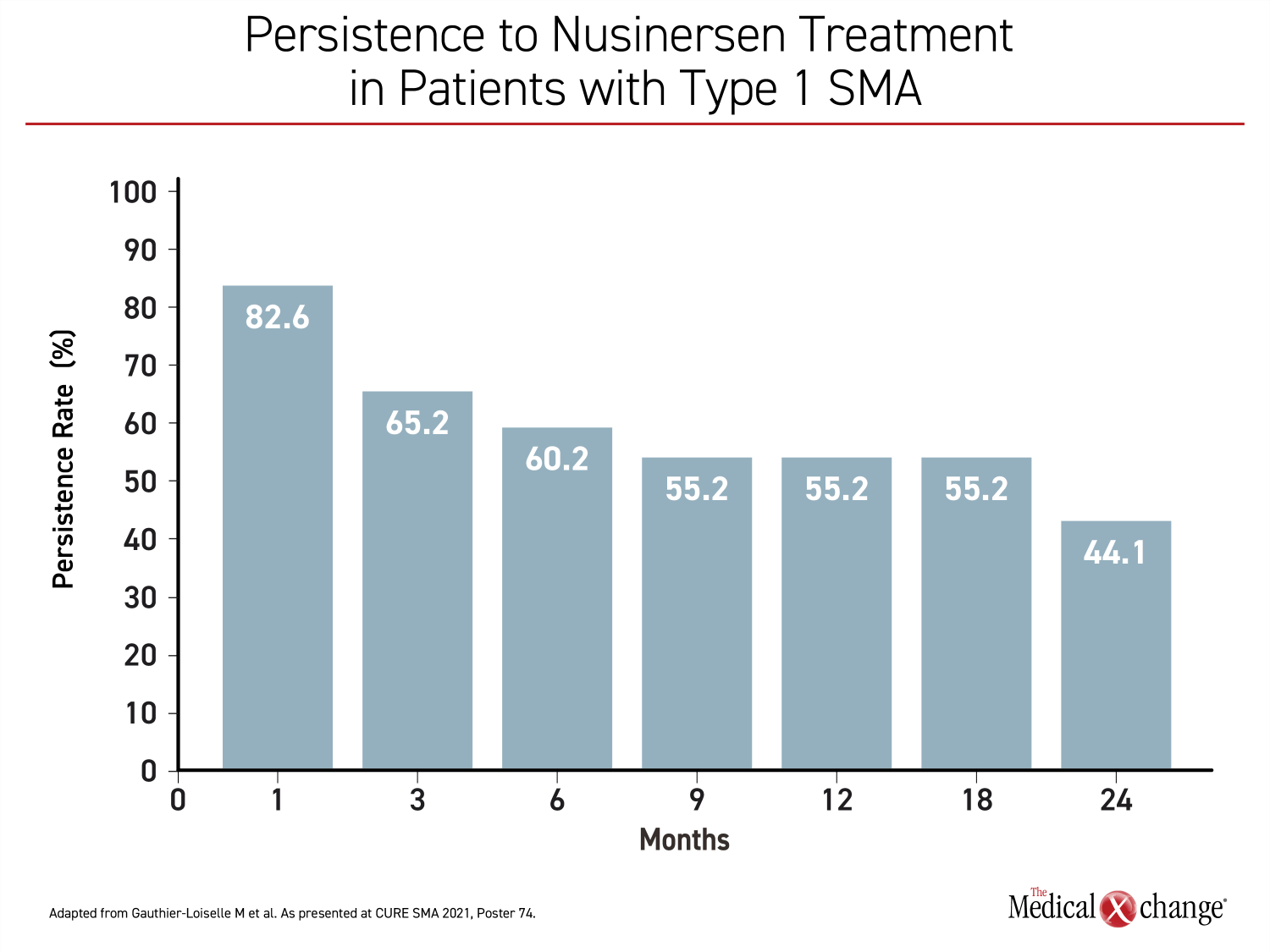
Treatment persistence did appear to be an issue based on a study of US nusinersen dosing data drawn from the Symphony Health Integrated Dataverseâ, which is a healthcare claims database. The objective was an analysis of real-world persistence on this therapy in patients with SMA types 1, 2, or 3. Over a two-year period of study, less than half of patients with type 1 SMA persisted on nusinersen (Figure 4). In the same group, which has the worst prognosis, 48% had discontinued therapy over the period of study, and the median time to discontinuation was 18.5 months. In types 2 and 3, discontinuation rates were higher, approaching 60%.
Patients who discontinued nusinersen, which was defined as two or more consecutive missed doses, “were more likely to have SMA comorbidities, such as feeding difficulties, dyspnea and respiratory anomalies, and muscle weakness,” reported Dr. Marjolaine Gauthier-Loiselle, Researcher and Manager, Analysis Group, Montreal, Quebec. She noted the increase in comorbidities was observed in type 2 and 3 SMA as well as type 1.
Treatment delays, defined as at least 28 days past the scheduled delivery of the maintenance intrathecal dose, were also common, occurring in about half of the patients. The reason for the discontinuations and delays could not be ascertained from these data, but Dr. Gauthier-Loiselle speculated that “logistical challenges,” such as travelling to treatment sites, might have contributed. She did not discuss how families perceive the inconvenience and discomfort of every-four-month intrathecal drug delivery, but she did caution that “treatment delays in this patient population may result in loss of motor neuron function and disease progression.”
“Treatment delays in this patient population may result in loss of motor neuron function and disease progression.”
WINDOW OF TIME FOR MILESTONES
In early childhood, there is evidence, including the experience generated with SMA treatments, that many neurological milestones must be achieved within a specific time window so as not to jeopardize gain of function. In a review of the LT-002 data, Dr. Kevin A. Straus, Medical director, Clinic for Special Children, Strasburg, Pennsylvania, reported that late treatment of SMA type 1 can provide major clinical benefits, but the greatest opportunity for age-appropriate gain of milestones appears to be derived from treatment before or in the earliest stages of symptoms.
“The strongest message of the data is to treat patients as early as possible to harness the benefit of disease-modifying therapies,” he said. This is relevant regardless of drug mechanism.
“The strongest message of the data is to treat patients as early as possible to harness the benefit of disease-modifying therapies.”
CONCLUSION
Therapies introduced for SMA have been revolutionary. For those with type 1 SMA, an inevitably fatal condition without treatment, the majority of patients treated in the first months of life with gene therapy have achieved age-appropriate milestones following a single treatment. Benefit from gene therapy appears to accrue even in older patients, including those with type 2 or 3 SMA, whether onasemnogene abeparvovec is the first or second therapy offered. The SMN2 gene splicing therapies are also associated with major improvements in outcome when patients remain adherent to the dosing schedules.