Expert Review
Severe Asthma: Characterization for Individualized Therapy
Chapter 1: Severe Asthma Phenotypes
Richard Leigh, MD, PhD
Professor of Medicine, University of Calgary,
Calgary, Alberta
It is now well accepted that the characteristic airway inflammation of asthma develops from multiple and distinct molecular processes. This heterogeneity is captured in the description of phenotypes, which are categorizations based on empirical observations. Differences in apparent triggers, age of onset, response to therapy, and other disease features support the hypothesis that the term asthma encompasses a set of clinical syndromes rather than a single disease entity. As an approach to diagnosis and treatment of severe asthma, phenotyping represents a fundamental reorientation to the presumption that a stepwise treatment algorithm can be uniformly applied to all asthma cases. In severe asthma poorly responsive to standard therapies, phenotyping has been the basis for understanding disease heterogeneity and to seek options for individualizing treatment. Most recently, phenotyping has been driving efforts to identify differences in immune modulators active in mediating airway inflammation. Progress in this area can be credited with a growing number of targeted therapies for severe disease.
Severe Asthma: Definition and Epidemiology
By guideline definition, asthma is severe when it is refractory to standard therapies. In the most recent European Respiratory Society/American Thoracic Society (ERS/ATS) guidelines, the specific criterion for severe asthma is persistent or recurrent uncontrolled symptoms over the previous year despite high doses of an inhaled corticosteroid (ICS) plus a long-acting beta agonist (LABA) or leukotriene modifier.1 Uncontrolled asthma without systemic corticosteroids maintained for at least 50% of the previous year is also an ERS/ATS criterion of severe disease. Uncontrolled asthma is defined as frequent exacerbations (≥2 bursts of systemic corticosteroids in previous year), serious exacerbations (≥1 hospitalization or intensive care unit stay in previous year), or airflow limitation (FEV1 <80% predicted) despite appropriate bronchodilator therapy (Fig. 1). It is estimated that about 10% of patients with asthma have severe disease.2 In Canada, which has an estimated asthma prevalence of 7% to 10%,3 as many as 250,000 individuals may have a severe form of asthma, according to data cited by Asthma Canada.4 The annual death toll from severe asthma in Canada is approximately 250 individuals (Fig. 2). Severe disease also accounts for a disproportionate proportion of asthma care costs. In a U.S. study, the cost of care over a 2-year period was twice as great in those with difficult-to-control than those with controlled asthma.5 The correlation between increasing asthma severity and diminishing quality of life is an unsurprising consequence of persistent symptoms and frequent office visits.6,7 A history of recurrent hospitalizations and need for ventilatory assistance represent risk factors for life-threatening episodes of asthma.8,9 Catastrophic episodes of asthma, including fatal events, are considered preventable by intensifying therapy, but severe asthma is not a single disease or itself a useful phenotype.10 Instead, the goals of grouping patients by phenotype is to gain insight into disease course, response to therapy, and, ultimately, underlying common pathobiology that may prove targetable. Variability in response to asthma therapies, particularly therapies targeted at the characteristic inflammation of asthma, provided the basis for the observation that events driving asthma pathophysiology are not uniform.11
Evolving Concepts of Asthma and Phenotyping
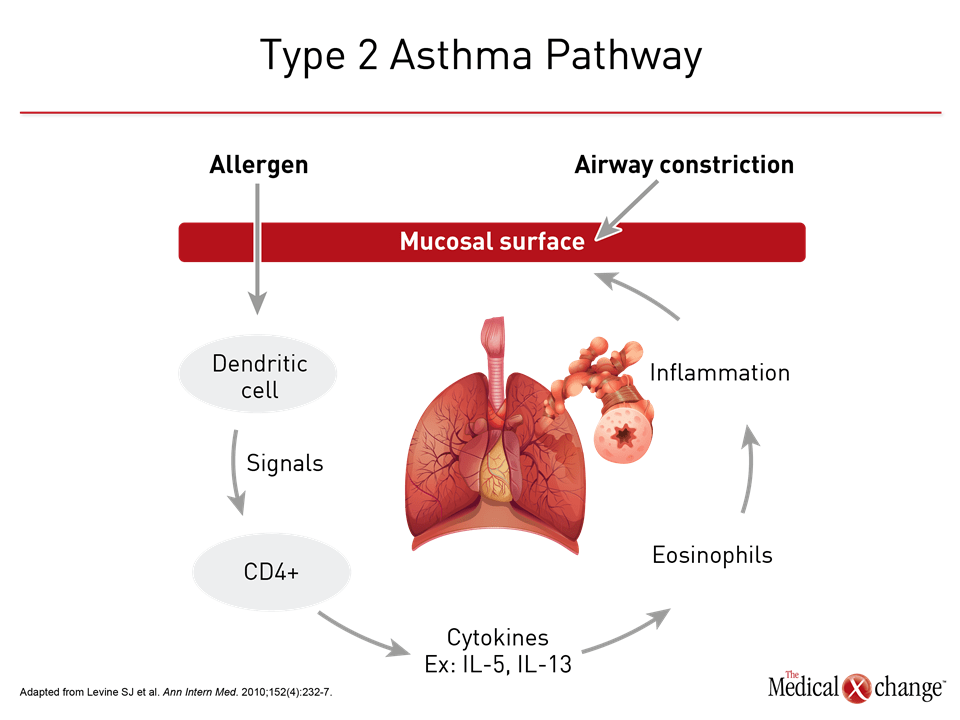
The effort to derive clinical relevance out of the heterogeneity of asthma dates back decades. Asthma subgroups were proposed on the basis of environmental triggers in 1947.12 In 1958, high and low sputum counts were identified as a potential tool for clinically relevant subtypes based on their association with likelihood of response to corticosteroids.13 Examples of subsequent subgroups or phenotypes proposed on observable characteristics included allergic versus non-allergic features,14 the presence of inflammatory cells based on biopsy,15 and clinical severity.16 As the number of features with potential clinical relevance increased, cluster analyses were applied in a data-centered approach to phenotyping. An initiative called the Severe Asthma Research Program (SARP), which compressed 34 variables in to 5 clinical asthma phenotypes, is a prominent example.17,18 Of efforts to phenotype asthma, there has been and continues to be a strong focus on the relative participation of inflammatory cells, particularly CD4+ T-helper 2 (Th2) lymphocytes, and their associated cytokines. While asthma was once considered to be an inflammatory process mediated primarily or exclusively by the Th2 cell pathway (Fig. 3), some patients have low expression of the cytokines associated with this type of immune response.19 Subsequently, two distinct molecular phenotypes were recognized. Initially referred to as Th2 high and Th2 low asthma, it is now understood that non-Th2 cells, such as mast cells, are also associated with upregulation of the classic Th2-associated cytokines, which include interleukin (IL)-4, IL-5, and IL-13.11 As a result, the alternative terminologies type 2 high or type 2 low asthma are sometimes employed in place of Th2. The key characteristics of type 2-high asthma include increased blood and airway eosinophilia, airway hyperresponsiveness, a thickened subepithelial basement membrane (SBM) and elevated IgE levels.20 The increase in eosinophils is attributed to type 2 mediated expression of IL-5 and IL-13 expression.21 Although type 2 high asthma is traditionally considered to be responsive to corticosteroids, severe asthma with eosinophilia is by definition poorly responsive to corticosteroids.22 Type 2 low asthma remains less well characterized. Although more closely associated with neutrophilic inflammation,10 type 2 low asthma does not exclude the expression of eosinophils. In a SARP program analysis, for example, four phenotypic clusters based on sputum neutrophils were identified including one with concurrent eosinophilia.23 Animal models have supported a role of Il-17 in neutrophlic inflammation,24 but it has also been hypothesized that neutrophilic inflammation in at least some patients with asthma is induced by extensive exposure to corticosteroids.11 Overall, a recent review concluded that no biomarkers for the type 2 low phenotype are yet considered to be clinically valid.25 Within type 2 high and type 2 low classifications, a large array of phenotypes can be derived from specific disease features, such as severity, age of onset, or association with environmental triggers, but not all asthma may fit within either of these immune response pathways. High IL-17 expression, for example, may represent a distinct pathway that is non-type 2 high or low but a product of upregulation of Th17 cells.26 Obesity, a risk factor for asthma, is another example. Factors such as chest wall biomechanics and airway compliance may contribute to the pathobiology of asthma in obese patients independent of immune response.27 For some patients, the key contributor to severe asthma may be corticosteroid resistance by one or more mechanisms, such as impaired glucocorticosteroid receptor binding.28
Phenotyping, Endotyping, and Genetics
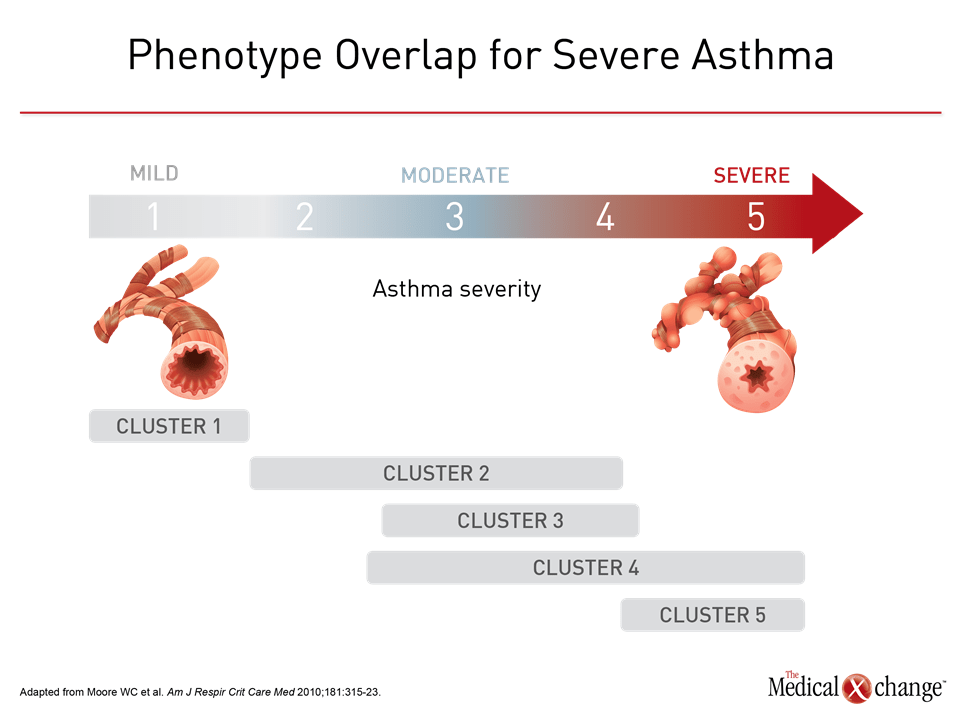
Phenotyping has been an empirical tool for capturing the heterogeneity of asthma, but the ultimate goal is to understand and treat the underlying molecular processes. This goal is particularly urgent in severe phenotypes defined by poor response to conventional therapies. As molecular mechanisms are defined, phenotypes have the potential to transform into endotypes, which describe subtypes of disease in which the molecular mechanisms are known. The efficacy of targeted therapies, such as omalizumab, which binds to IgE, and the more recently licensed anti-IL-5 agents have specific molecular targets, but do not yet have well defined asthma endotypes for which efficacy or lack of efficacy is absolutely or even strongly correlated with the presence of absence of the putative molecular targets. In these cases, it appears that putative targets may be necessary but not sufficient to predict response. In placebo-controlled trials with omalizumab, for example, post-hoc analyses suggest that patients with a history of allergic asthma with IgE levels >75 IU/mL have a lower annualized exacerbation rate than in those with lower IgE levels, but there is no dose response for levels above this threshold, and other features, such as poor previous response to relatively high doses of corticosteroids, are predictors of response independent of the molecular target.29 Omalizumab is effective relative to placebo in patients with moderate to severe persistent asthma who have a positive skin test or in vitro reactivity to a perennial aeroallergen, but it is typically offered as an adjunct to other asthma treatments to the limited control achieved when this treatment is used alone.30 The targeted anti-IL-5 therapies appear to be more specific. In initial studies with mepolizumab, there was no benefit relative to placebo in unselected patients with asthma inadequately responsive to corticosteroids.31 Subsequent trials with this drug and with two other anti IL-5 therapies, reslizumab and benralizumab, did show significant protection against exacerbations when eosinophilia was a trial entry criterion.32-34 While IL-5 has proven to be an important biomarker in identifying patients most likely to benefit from these targeted agents, not all patients with eosinophilia respond to this medication, suggesting that patients have additional mechanisms participating in disease expression. One obstacle to defining asthma phenotypes on the path to identification of treatable endotypes is complexity. The large number of attempts to phenotype asthma reveals overlapping clinical and molecular features suggesting that asthma in many or most patients is the product of several mechanisms.35 In a study of over 500 adults and children, more than half of patients fell into two or more phenotypes defined by atopic, eosinophilic, and type 2 asthma criteria.36 The rates of overlap depended on how these phenotypes were defined. For example 31% to 78% of children and 21% to 69% of adults fell into the eosinophilic phenotype depending on whether eosinophil cutoffs of ≥150, ≥300, or ≥450 eosinophils/μL were employed. Numerous other studies have reported on the frequency of phenotype overlaps.37,38 In a study from SARP, Moore and colleagues used unsupervised hierarchical cluster analysis to identify five distinct, but overlapping asthma phenotypes; subjects in Cluster 1 have early onset atopic asthma with normal lung function treated with two or fewer controller medications and minimal health care utilization.18 Subjects in Cluster 2 have early-onset atopic asthma and preserved lung function, but increased medication requirements and health care utilization. Cluster 3 comprises mostly older obese women with late-onset nonatopic asthma, moderate reductions in FEV1, and frequent oral corticosteroid use to manage exacerbations. Subjects in Clusters 4 and 5 have severe airflow obstruction with variable bronchodilator responsiveness, but differ in to their ability to attain normal lung function, age of asthma onset, atopic status, and use of oral corticosteroids. Except for clusters 1 and 5, there was substantial overlap for asthma severity (Fig. 4). Phenotype overlap has been further supported by a gene set analysis from sputum cell transcriptomics based on samples from 104 patients with moderate to severe asthma.39 Three distinct molecular phenotypes were derived from the hierarchical clustering. Eosinophilic asthma was dominant in one of these clusters but it was not exclusive to this cluster. Rather, it was also significantly present in a second cluster, while two clusters that were consider non type 2 phenotypes were associated with expression of interferon and tumor necrosis factor cytokines. Although only a single study in a limited study population, the findings emphasize the potential complexity of molecular signaling underlying asthma severity. The vast array of phenotypes created by clinical and molecular characteristics, the evidence of considerable phenotype overlap, and the potential for phenotypes to shift as patients progress from mild to severe asthma explain some the complexity of applying phenotyping in clinical disease management. Although there is a strong consensus that phenotyping or cluster analysis will eventually prove to be a useful tool in personalized care of asthma,17 the current application has been primarily as a research tool.40
Conclusion
Phenotypes provide a framework with which to explore patterns within the heterogeneity of factors that drive severe asthma. These phenotypes, defined variably, provide compelling evidence that asthma is not a single disease but the final expression of multiple pathological processes. For patients with severe airway inflammation, this direction of research has provided a framework for understanding underlying molecular events for disease expression and is leading toward increasingly personalized therapy for disease control.
References
- Chung KF, Wenzel SE, Brozek JL, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J 2014;43:343-73.
- Hekking PP, Wener RR, Amelink M, Zwinderman AH, Bouvy ML, Bel EH. The prevalence of severe refractory asthma. J Allergy Clin Immunol 2015;135:896-902.
- Rowe BH, Villa-Roel C, Abu-Laban RB, et al. Admissions to Canadian hospitals for acute asthma: a prospective, multicentre study. Can Respir J 2010;17:25-30.
- Asthma Facts and Statistics. Asthma Canada. Toronto, Canada: https://asthma.ca/wp-content/uploads/2017/06/Asthma-Facts-and-Stats.pdf; Accessed, August 9, 2017.
- Sullivan SD, Rasouliyan L, Russo PA, Kamath T, Chipps BE, Group TS. Extent, patterns, and burden of uncontrolled disease in severe or difficult-to-treat asthma. Allergy 2007;62:126-33.
- Siroux V, Boudier A, Anto JM, et al. Quality-of-life and asthma-severity in general population asthmatics: results of the ECRHS II study. Allergy 2008;63:547-54.
- Gonzalez-Barcala FJ, de la Fuente-Cid R, Tafalla M, Nuevo J, Caamano-Isorna F. Factors associated with health-related quality of life in adults with asthma. A cross-sectional study. Multidiscip Respir Med 2012;7:32.
- McFadden ER, Jr., Warren EL. Observations on asthma mortality. Ann Intern Med 1997;127:142-7.
- Omachi TA, Iribarren C, Sarkar U, et al. Risk factors for death in adults with severe asthma. Ann Allergy Asthma Immunol 2008;101:130-6.
- Ray A, Raundhal M, Oriss TB, Ray P, Wenzel SE. Current concepts of severe asthma. J Clin Invest 2016;126:2394-403.
- Gauthier M, Ray A, Wenzel SE. Evolving Concepts of Asthma. Am J Respir Crit Care Med 2015;192:660-8.
- Rackemann FM. A working classification of asthma. Am J Med 1947;3:601-6.
- Brown HM. Treatment of chronic asthma with prednisolone; significance of eosinophils in the sputum. Lancet 1958;2:1245-7.
- Walker C, Virchow JC, Jr., Bruijnzeel PL, Blaser K. T cell subsets and their soluble products regulate eosinophilia in allergic and nonallergic asthma. J Immunol 1991;146:1829-35.
- Wenzel SE, Schwartz LB, Langmack EL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med 1999;160:1001-8.
- Miranda C, Busacker A, Balzar S, Trudeau J, Wenzel SE. Distinguishing severe asthma phenotypes: role of age at onset and eosinophilic inflammation. J Allergy Clin Immunol 2004;113:101-8.
- Haldar P, Pavord ID, Shaw DE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med 2008;178:218-24.
- Moore WC, Meyers DA, Wenzel SE, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med 2010;181:315-23.
- Fahy JV. Type 2 inflammation in asthma–present in most, absent in many. Nat Rev Immunol 2015;15:57-65.
- Woodruff PG, Modrek B, Choy DF, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med 2009;180:388-95.
- Pope SM, Brandt EB, Mishra A, et al. IL-13 induces eosinophil recruitment into the lung by an IL-5- and eotaxin-dependent mechanism. J Allergy Clin Immunol 2001;108:594-601.
- Buhl R, Humbert M, Bjermer L, et al. Severe eosinophilic asthma: a roadmap to consensus. Eur Respir J 2017;49.
- Moore WC, Hastie AT, Li X, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol 2014;133:1557-63 e5.
- Lajoie S, Lewkowich IP, Suzuki Y, et al. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol 2010;11:928-35.
- Robinson D, Humbert M, Buhl R, et al. Revisiting Type 2-high and Type 2-low airway inflammation in asthma: current knowledge and therapeutic implications. Clin Exp Allergy 2017;47:161-75.
- Busse WW, Holgate S, Kerwin E, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med 2013;188:1294-302.
- Al-Alwan A, Bates JH, Chapman DG, et al. The nonallergic asthma of obesity. A matter of distal lung compliance. Am J Respir Crit Care Med 2014;189:1494-502.
- Leung DY, Martin RJ, Szefler SJ, et al. Dysregulation of interleukin 4, interleukin 5, and interferon gamma gene expression in steroid-resistant asthma. J Exp Med 1995;181:33-40.
- Chapman KR, Cartier A, Hebert J, McIvor RA, Schellenberg RR. The role of omalizumab in the treatment of severe allergic asthma. Can Respir J 2006;13 Suppl B:1B-9B.
- Holgate ST, Chuchalin AG, Hebert J, et al. Efficacy and safety of a recombinant anti-immunoglobulin E antibody (omalizumab) in severe allergic asthma. Clin Exp Allergy 2004;34:632-8.
- Flood-Page P, Swenson C, Faiferman I, et al. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med 2007;176:1062-71.
- Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet 2012;380:651-9.
- Castro M, Zangrilli J, Wechsler ME, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med 2015;3:355-66.
- FitzGerald JM, Bleecker ER, Nair P, et al. Benralizumab, an anti-interleukin-5 receptor alpha monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2016;388:2128-41.
- Papi A, Saetta M, Fabbri L. Severe asthma: phenotyping to endotyping or vice versa? Eur Respir J 2017;49.
- Tran TN, Zeiger RS, Peters SP, et al. Overlap of atopic, eosinophilic, and TH2-high asthma phenotypes in a general population with current asthma. Ann Allergy Asthma Immunol 2016;116:37-42.
- Loza MJ, Djukanovic R, Chung KF, et al. Validated and longitudinally stable asthma phenotypes based on cluster analysis of the ADEPT study. Respir Res 2016;17:165.
- Gaga M, Brand PL, Thomson NC. The quest for the grail: multidimensional efforts for understanding and targeting severe asthma. Eur Respir J 2015;46:1227-31.
- Kuo CS, Pavlidis S, Loza M, et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J 2017;49.
- Desai M, Oppenheimer J. Elucidating asthma phenotypes and endotypes: progress towards personalized medicine. Ann Allergy Asthma Immunol 2016;116:394-401.
Chapter 1: Severe Asthma Phenotypes
It is now well accepted that the characteristic airway inflammation of asthma develops from multiple and distinct molecular processes. This heterogeneity is captured in the description of phenotypes, which are categorizations based on empirical observations. Differences in apparent triggers, age of onset, response to therapy, and other disease features support the hypothesis that the term asthma encompasses a set of clinical syndromes rather than a single disease entity. As an approach to diagnosis and treatment of severe asthma, phenotyping represents a fundamental reorientation to the presumption that a stepwise treatment algorithm can be uniformly applied to all asthma cases. In severe asthma poorly responsive to standard therapies, phenotyping has been the basis for understanding disease heterogeneity and to seek options for individualizing treatment. Most recently, phenotyping has been driving efforts to identify differences in immune modulators active in mediating airway inflammation. Progress in this area can be credited with a growing number of targeted therapies for severe disease.
Show review