Neurology
7th Congress of the European Academy of Neurology (EAN) - Virtual 2021
STR1VE-EU: Gene Replacement Therapy in Symptomatic Spinal Muscular Atrophy Is Associated with Substantial Benefit
Virtual Meeting – For type 1 spinal muscular atrophy (SMA), new data associate gene replacement therapy with important benefits even when administered after symptoms have developed. The phase 3 study included a substantial proportion of patients who required respiratory support, nutritional support, or both at the time that they received this therapy. Of the 33 children enrolled, there was at least some degree of clinical improvement in all. Of the 32 evaluable patients, 31 (97%) were alive and free of permanent ventilation at 18 months of age. All children achieved developmental motor milestones that would not have been possible without therapy.
In previous studies with this gene replacement therapy, called onasemnogene abeparvovec, most patients achieved and sustained developmental milestones when a single infusion was delivered within weeks of birth. The results of this phase 3 study, called STR1VE-EU, indicate that a large clinical benefit can be obtained even when treatment is delayed. The results of this European trial are similar to those from a somewhat smaller and recently published 12-center US trial called STR1VE-US (Day JW et al. Lancet Neurol 2021;20:284-293), but the patients in STR1VE-EU tended to be sicker at entry.
“STR1VE-EU associated onasemnogene abeparvovec with strong efficacy in terms of survival and functional improvement that included improvement even in those with advanced disease,” reported Dr. Eugenio Mercuri, Department of Pediatric Neurology, Catholic University, Rome, Italy. He called the benefit-to-risk profile “remarkably favourable in this type 1 symptomatic population.”
“STR1VE-EU associated [the gene replacement therapy] with strong efficacy in terms of survival and functional improvement.”
Type 1 SMA Typically Fatal
All patients enrolled in STR1VE-EU had type 1 SMA, the most common and most severe form of this disease. Like all SMA, it is characterized by loss-of-function mutations in SMN1, a gene responsible for generating the SMN1 protein essential for neuromuscular development. Type 1 patients have no more than two copies of SMN2, which also can produce SMN protein. Unlike types 2 and 3 SMA, which have more copies of the SMN2 gene, type 1 SMA patients rarely survive to the age of 24 months without treatment.
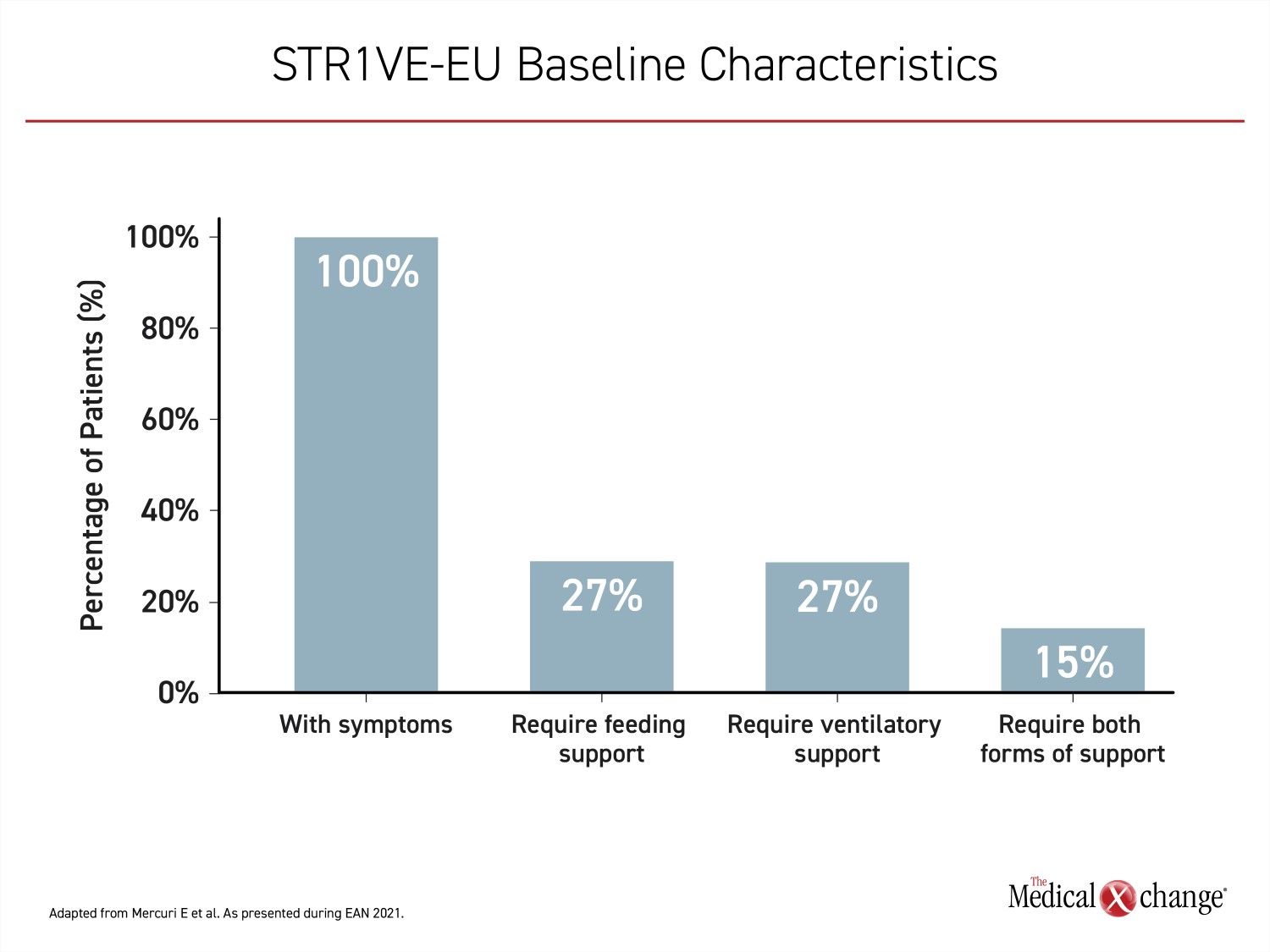
In STR1VE-EU, patients were required to be enrolled before six months of age and the median age at time of treatment was 4.1 months. By 3 months of age, which was the earliest age of treatment initiation in this study, all the infants were symptomatic. The mean age of symptom onset was 1.5 months. When enrolled, nine (27%) required ventilatory support, nine (27%) required nutritional support, and five (15%) required both. Twenty patients had not yet advanced to a severity requiring these forms of life support (Figure 1).
The primary endpoint for this study was the ability to sit independently for at least 10 seconds by the age of 18 months. The secondary endpoint was survival without respiratory support. The array of exploratory endpoints included change in motor function as captured with the Children’s Hospital of Philadelphia Infant Test of Neurological Disorders (CHOP INTEND) and Bayley-III motor milestones. Outcomes of enrolled patients were compared to the Pediatric Neuromuscular Clinical Research Network (PNCR) cohort of untreated type 1 SMA patients.
44% Achieve Primary Endpoint
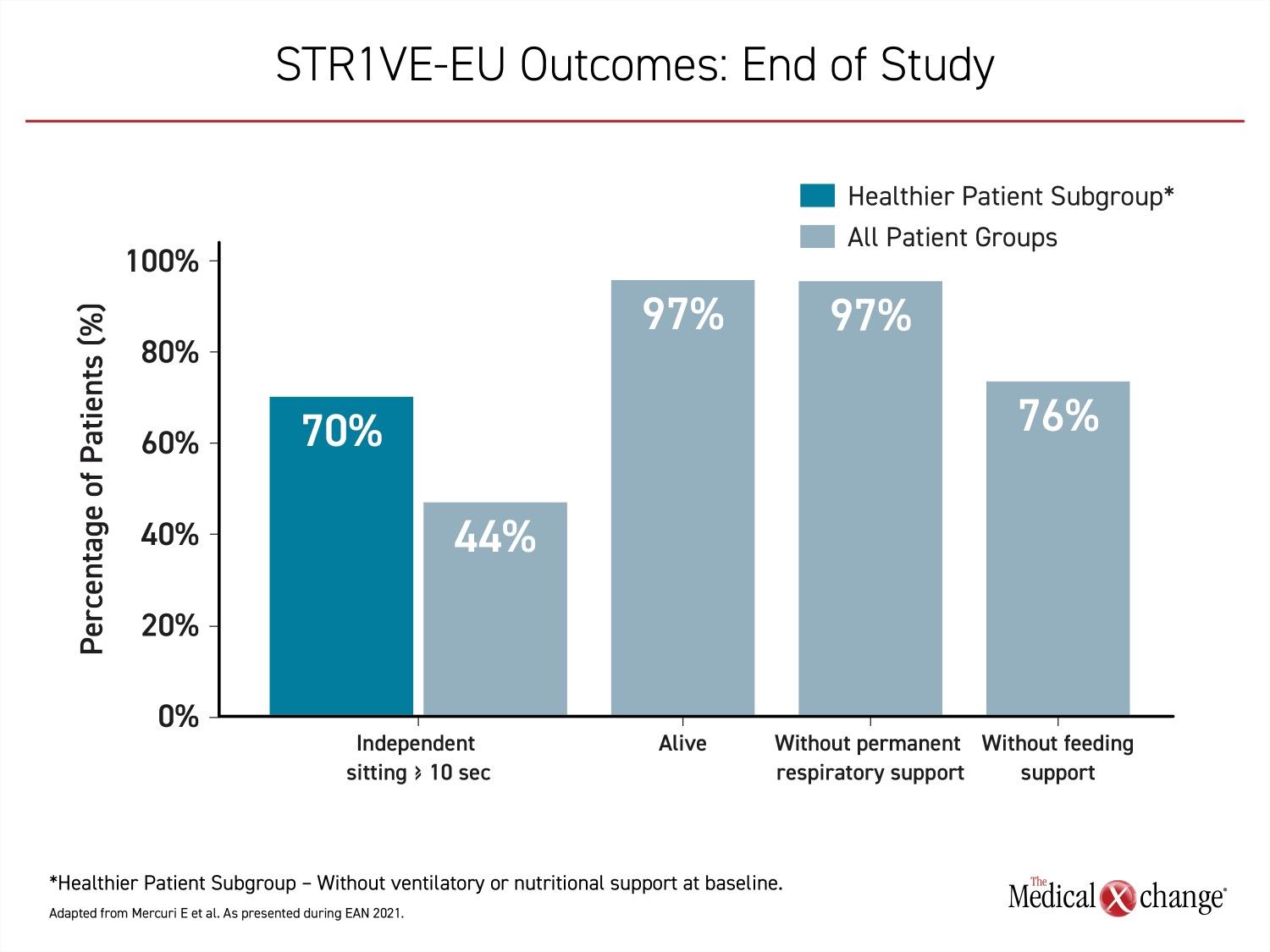
The primary endpoint of functional independent sitting for ≥10 seconds, as defined by the World Health Organization (WHO) was achieved in 14 (44%) of the 32 evaluable patients. The median age for those who achieved this milestone was 15.9 months. The youngest age was 7.7 months. In the subgroup of infants who did not require ventilatory or respiratory support at baseline, the primary endpoint was reached by 70% (Figure 2).
Greater delay in treatment did not necessarily predict lower likelihood of achieving the primary endpoint. Dr. Mercuri noted that one of the infants enrolled and subsequently excluded from the final analysis, because gene replacement therapy was administered after the six-month-of-age entry criterion, still achieved the primary endpoint.
In a PNCR cohort of untreated type 1 SMA patients that was used for reference, no child ever achieved independent sitting prior to death. In STR1VE-EU, there was a single death over the course of follow-up, which was determined to be unrelated to treatment. This 97% rate of survival compares to no survival in the PNCR cohort (P<0.0001). None of the 32 evaluable patients who survived until at least 18 months of age required permanent ventilatory support. Of the nine patients on non-invasive ventilatory support at baseline, two were fully independent of respiratory support by the end of follow-up. Of the 24 patients who were not on ventilatory support at baseline, 16 remained free of any kind respiratory assistance.
Steady Increase in CHOP INTEND and Bayley-III
The median CHOP INTEND score at study entry was 28.0, which represents significant deficits in motor function. The lowest score was 22.0. After administration of onasemnogene abeparvovec, scores began climbing at least modestly in all patients and steeply in most. The mean increases were 6.0 points at one month, 10.3 points at month 3, and 13.8 points at month 6.
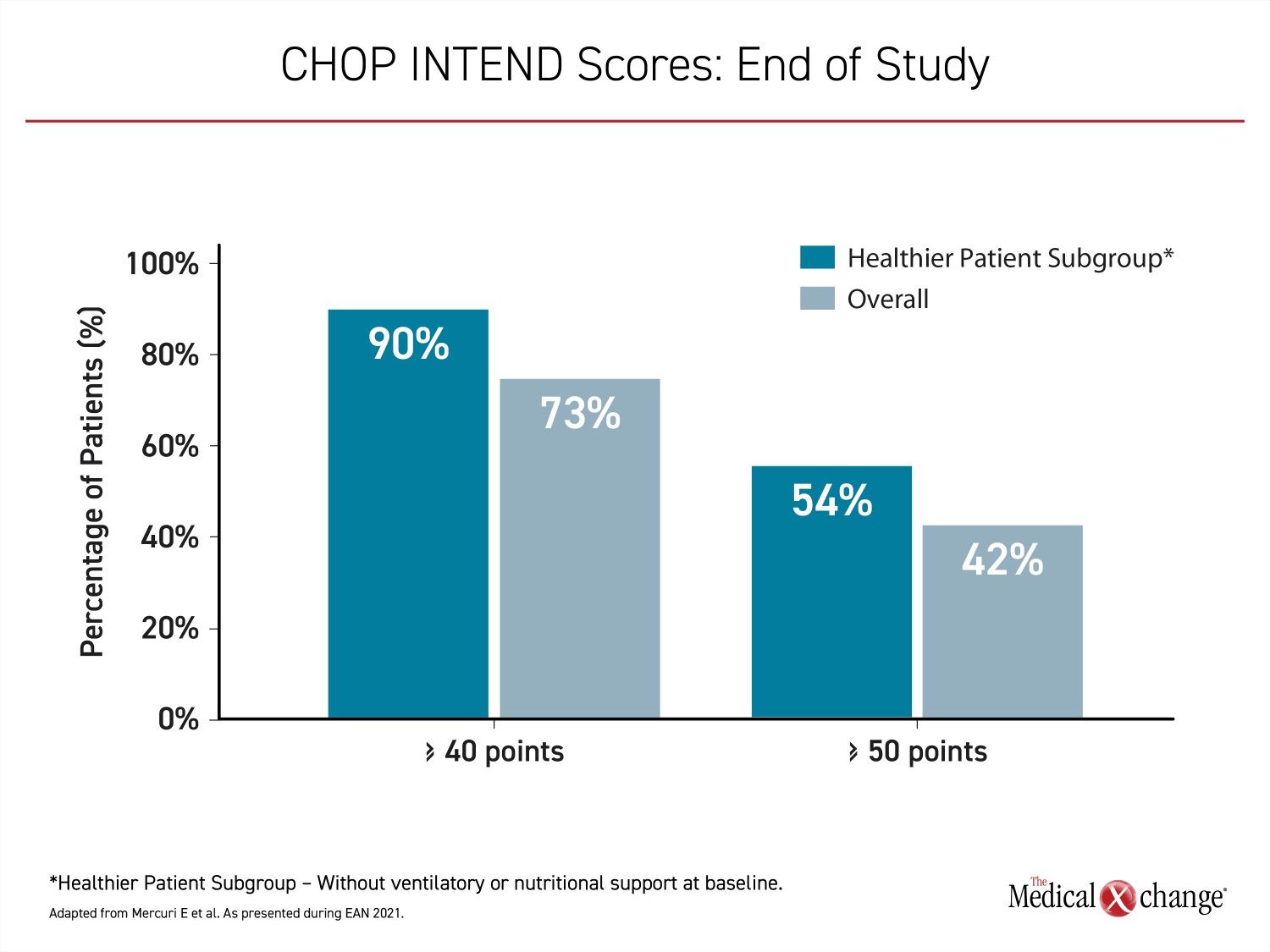
“At the end of the study, 24 patients (73%) achieved CHOP INTEND scores of 40 or greater, 14 patients (42%) achieved scores of 50 points or great, and three (9%) patients achieved a CHOP INTEND score of at least 58 points,” Dr. Mercuri reported. “These CHOP INTEND scores are remarkable in that patients with untreated SMA type 1 almost never achieve a CHOP INTEND score greater than 40 points.”
“These CHOP INTEND scores are remarkable in that patients with untreated SMA type 1 almost never achieve a CHOP INTEND score greater than 40 points.”
Children that did not require ventilatory or nutritional support at baseline achieved even greater gains in CHOP INTEND scores over the course of the study (Figure 3).
Of the 30 infants without head control at baseline, 27 (90%) had achieved this milestone by the end of the study. As defined by the Bayley-III motor milestones, 19 patients (56%) achieved the ability to roll from back to side. Sixteen patients (49%) demonstrated the ability to sit unassisted for at least 30 seconds.
>75% Free of Nutritional Support
At the end of the study, eight patients required some form of nutritional support. Of these, four were among the nine children who required nutritional support at baseline. The remaining five had gained the ability to feed without mechanical support. While four patients who had not needed nutritional support at baseline did at the end of the study, 25 (78%) of the 32 evaluable patients did not.
No New Safety Issues with Gene Replacement Therapy
Hepatotoxicity was the most common adverse event likely to be treatment related. Twenty-two patients (63.7%) had elevated liver enzymes sometime during the study. Fibrosis, cirrhosis, and other signs of liver damage were observed in 18 patients (54.5%). However, hepatoxicity was generally asymptomatic and self-resolved or responded to prednisone.
According to Dr. Mercuri, the safety profile was consistent with that observed in other trials with onasemnogene abeparvovec including the STR1VE-US study. In that study, two of the three serious adverse events possibly related to onasemnogene abeparvovec involved elevations of hepatic aminotransferases. The other was a case of hydrocephalus. All resolved.
“We did not see any new safety signals,” Dr. Mercuri said. Other than elevated liver enzymes, the most frequently reported adverse events were pyrexia and respiratory infections. These events, which are common in infants, were not clearly treatment related.
STR1VE-US Show Similar Benefits
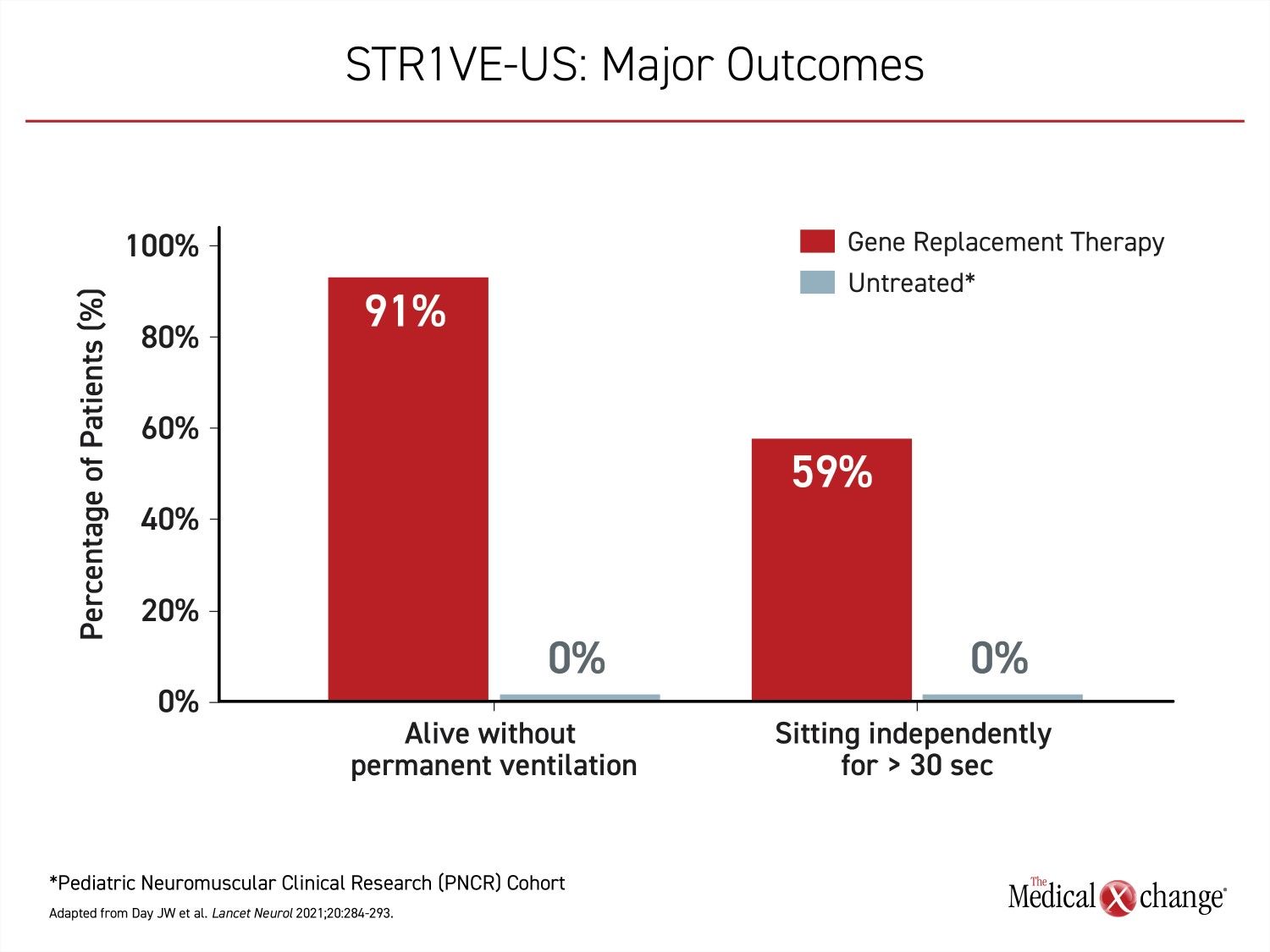
STR1VE-US enrolled 22 symptomatic type 1 SMA patients, all of which received onasemnogene abeparvovec before six months of age. The primary endpoint in this study was functional independent sitting for at least 30 seconds before 18 months of age, and was achieved by 13 (59%) of patients. Relative to the PNCR cohort, in which this milestone was never achieved, the difference in STR1VE-US, like STR1VE-EU, was highly significant P<0.0001) (Figure 4).
STR1VE-EU Reproduces STR1VE-US Results
Similar to STR1VE-EU, 20 (91%) of the patients in STR1VE-US survived free from permanent ventilation, confirming a major benefit from gene replacement therapy in symptomatic type 1 SMA patients. Although these studies do not challenge the goal of treating type 1 SMA with gene replacement therapy as quickly as possible after the diagnosis, it does support treatment even if the diagnosis is delayed.
“Those with better baseline respiratory function had much higher scores than those who had some ventilatory deficit. However, these [patients with impaired respiratory function] still showed improvement even if the absolute improvement was lower,” Dr. Mercuri said.
“Patients with impaired respiratory function at baseline still showed improvement even if the absolute improvement was lower.”
There are no published systematic data assessing the clinical value of onasemnogene abeparvovec in children with type 1 SMA who are more than six months of age, but the RESTORE Registry is now following more than 45 patients with type 1 SMA who received gene therapy at the age of six months or older. As in the phase 3 STR1VE trials, symptom improvement has been reported. Although it is believed that at least some developmental milestones are irretrievably lost if restoration of SMN1 protein is not provided within or prior to the window in which specific neurodevelopment is expected, these data support treatment even after type 1 SMA children have become symptomatic.
Gene Replacement Therapy Prior to Symptoms Is Ideal
If onasemnogene abeparvovec is administered within the first few weeks of life and before the onset of symptoms, most children achieve major early development milestones. In the phase 3 SPR1NT trial, which was presented as a latebreaker at the 2021 EAN Congress, all 14 of the patients in the type 1 SMA, two-copy SMN2 cohort met the primary endpoint of sitting without support for at least 30 seconds. Most did so during the normal expected window that this milestone is achieved.
None of the patients in this study “have required any form of nutritional or ventilatory support” so far, according to Dr. Kevin A. Strauss, Medical Director, Clinic for Special Children, Strasburg, Pennsylvania.
“None of the patients in [SPR1NT] have required any form of nutritional or ventilatory support.”
Even though the follow-up has not exceeded the window of time for other expected milestones, 79% of the children treated when asymptomatic are now standing without support for at least three seconds, and 61% are walking, Dr. Strauss reported.
Overall, these data “lend further support for the importance of early diagnosis of SMA,” said Dr. Strauss who advocates universal pre- or perinatal screening to accelerate the time to diagnosis.
In SMA type 1 patients treated soon after birth, at least some infants have achieved normal and sustained neurodevelopment through follow-up to date. Among infants participating in the phase 3 study with onasemnogene abeparvovec, some have now been followed for more than 5.5 years.
Conclusion
The final results of the STR1VE-EU trial, consistent with the previously published STR1VE-US study, demonstrate that there are significant benefits from the gene replacement therapy onasemnogene abeparvovec even when treatment is initiated in infants with symptomatic type 1 SMA. In both studies, children achieved important developmental milestones after a single infusion. Due to the somewhat greater severity of symptoms at baseline relative to the previously completed STR1VE-US study, STR1VE-EU provides substantial support for gene replacement therapy even if delayed beyond the ideal window of administration.