Neurology
World Muscle Society (WMS) 2021 Virtual Congress
Gene Replacement Therapy for SMA is Effective in Both Pre-symptomatic Infants and Older Symptomatic Children
Virtual Meeting – There is new evidence that a gene replacement therapy highly effective in the treatment of spinal muscular atrophy (SMA) when administered pre-symptomatically is also effective if initiated after symptoms have already developed. In data presented at the WMS 2021 Virtual Congress, substantial efficacy was documented in older children (≥6 months), whether or not they had received a prior disease-modifying therapy. While it is clear that gene replacement therapy is best delivered within weeks of birth and prior to the onset of symptoms, a delay in diagnosis and treatment initiation does not preclude benefit.
There are now three therapies approved for SMA in Canada. The first therapy, the antisense oligonucleotide nusinersen, was joined by a gene replacement therapy, onasemnogene abeparvovec, in late 2020. Risdiplam a small molecule, was more recently approved and like nusinersen, it also is a SMN2 splicing modifier that transiently impacts protein production and therefore requires continual maintenance dosing to prevent SMA progression. In comparison, gene replacement therapy replaces the SMN1 gene encoding the SMN protein, the defining deficit of SMA. In most patients, it offers indefinite benefit after a single dose.
“Deletion of the SMN1 gene is associated with loss of both voluntary motor function and bulbar function, which is essential for breathing and swallowing.”
“The deletion of the SMN1 gene is associated with the loss of both voluntary motor function and bulbar function, which is essential for breathing and swallowing. All of the disease-modifying treatments profoundly improve prognosis but differ in duration of benefit,” according to Dr. Laurent Servais, Professor of Pediatric Neuromuscular Diseases, University of Oxford, UK.
Benefit of Gene Replacement Therapy Studied in Older Children
In the development of the disease-modifying therapies, nusinersen and risdiplam were tested in infants and older children. In trials with onasemnogene abeparvovec, most children were less than 6 months of age at enrollment and had type 1 SMA, the most common form. Unlike SMA types 2 and 3, which might not produce symptoms for months or years, SMA type 1 typically produces symptoms within weeks of birth and is uniformly fatal without treatment. The SMA therapies are active against all types, but the absence of trial data with gene replacement therapy in older children created a knowledge gap and the “need for real-world data in a group of children who also urgently require treatment,” according to Dr. Servais.
Two sets of data presented here addressed this knowledge gap. The first, drawn from the multinational prospective RESTORE registry, was presented by Dr. Servais. The other compared health care resource utilization (HCRU) for children ≥6 months of age initiated on nusinersen or gene replacement therapy. Both sets of data supported the efficacy of gene replacement therapy in older children whether initiated as first line or after another disease-modifying therapy.
In the RESTORE Registry, more than 250 children have been enrolled so far. These patients have been recruited from all settings, not just clinical studies. Of these, 117 patients were ≥6 months of age when they were treated with onasemnogene abeparvovec. Most (66.7%) were diagnosed with type 1 SMA. Of the remainder, approximately 10% were presymptomatic.
RESTORE Registry: Benefits of Therapy Tested in Two Older Age Groups
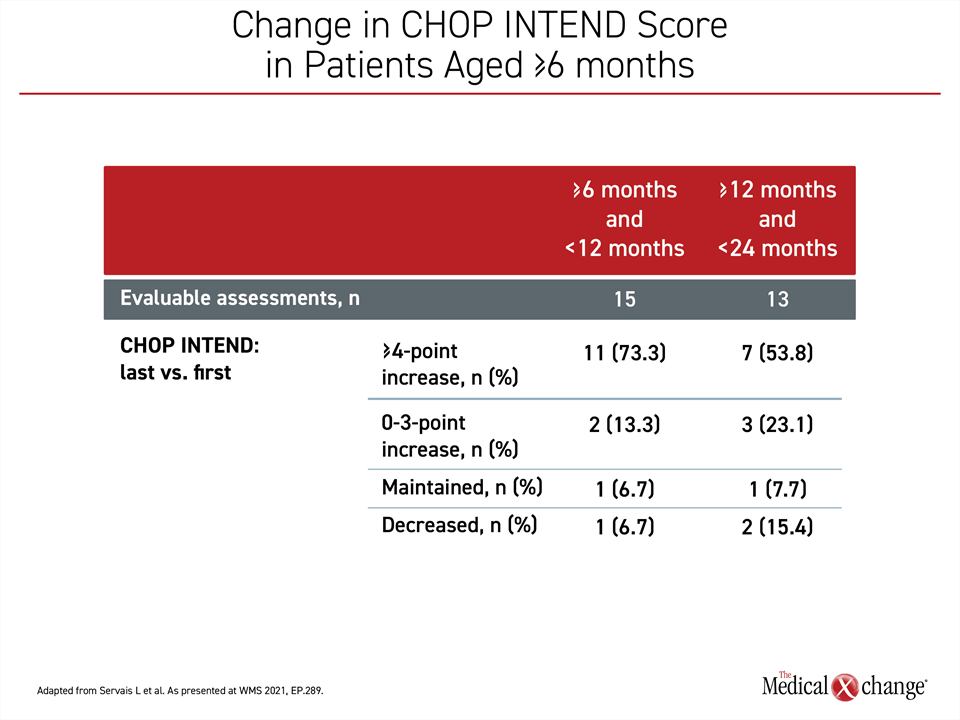
The assessment of benefit from a one-time infusion of gene replacement therapy in older children was measured with change in the objective functional scoring system CHOP INTEND (Children’s Hospital of Philadelphia Infant Test of Neurological Disorders). The patients were evaluated with CHOP INTEND at baseline and then at least once over the course of follow up. Analysis was stratified into children receiving gene replacement therapy between ≥6 to <12 months (n=15), those treated between ≥12 to <24 months (n=13), and those treated ≥24 months of age. The two younger groups were divided nearly evenly. The smaller proportion treated after 24 months of age (7.5%) was too small to assess separately.
Of children with evaluable motor function assessments following gene replacement therapy, the CHOP INTEND scores at baseline were compared to those at the most recent follow-up. Overall, these scores increased by ≥4 points in 73.3% of the children who received gene replacement therapy at ≥6 months of age but <12 months of age. The score declined in only one of the 15 patients in this group. Of the others, one maintained the pre-treatment CHOP INTEND score and the others had an increase of 1 to 3 points. Of children who received gene replacement therapy by ≥12 months but <24 months of age, the CHOP INTEND score increased by ≥4 points in 53.8%. Two of the 13 children in this group had a decline in score. One child maintained the pre-treatment CHOP INTEND score, and the remaining patients increased their score by 1 to 3 points (Figure 1).
Gene Replacement Therapy Safety Evaluated After Prior Therapy
In children ≥6 months of age but <12 months, the proportions treated with gene replacement therapy first line, after another disease-modifying therapy, or in combination with another disease-modifying therapy were similar. In children ≥12 months of age, only 29.8% received gene replacement therapy first line while 54.4% were treated with gene replacement therapy after another disease-modifying therapy. The remaining 15.8% received a combination of disease-modifying therapies. Prior exposure to SMA therapy did not appear to affect response rates to gene replacement therapy although the sample sizes for comparison were small, according to Dr. Servais.
Overall, rates of treatment-emergent adverse events in these older patients were similar to those previously reported in the pivotal trials with gene replacement therapy. However, the risk of events potentially treatment related might have been slightly higher in children between the ages of 12 and 24 months when compared to the younger age group. This included hepatotoxicity (43.9% vs. 34.0%), which consisted mostly of liver enzyme elevations, and transient thrombocytopenia (26.3% vs. 16.0%). There were no dorsal root ganglion toxicities reported in any patient, and there were low rates of symptomatic adverse events.
[Gene replacement therapy] “is an effective treatment choice for children with SMA ≥6 months of age regardless of prior disease-modifying therapy.”
Dr. Servais called the ≥4-point increase in CHOP INTEND, which was achieved in the majority of older children, “clinically meaningful.” In addressing the knowledge gap for use of gene replacement therapy in older children, he described these data as reassuring.
“Onasemnogene is an effective treatment choice for children with SMA ≥6 months of age regardless of prior disease-modifying therapy,” Dr. Servais reported. He added that the safety profile “is consistent” with the safety and tolerability previously reported in the pivotal trials.
Lower SMA-related HCRU Outcomes in Older Children
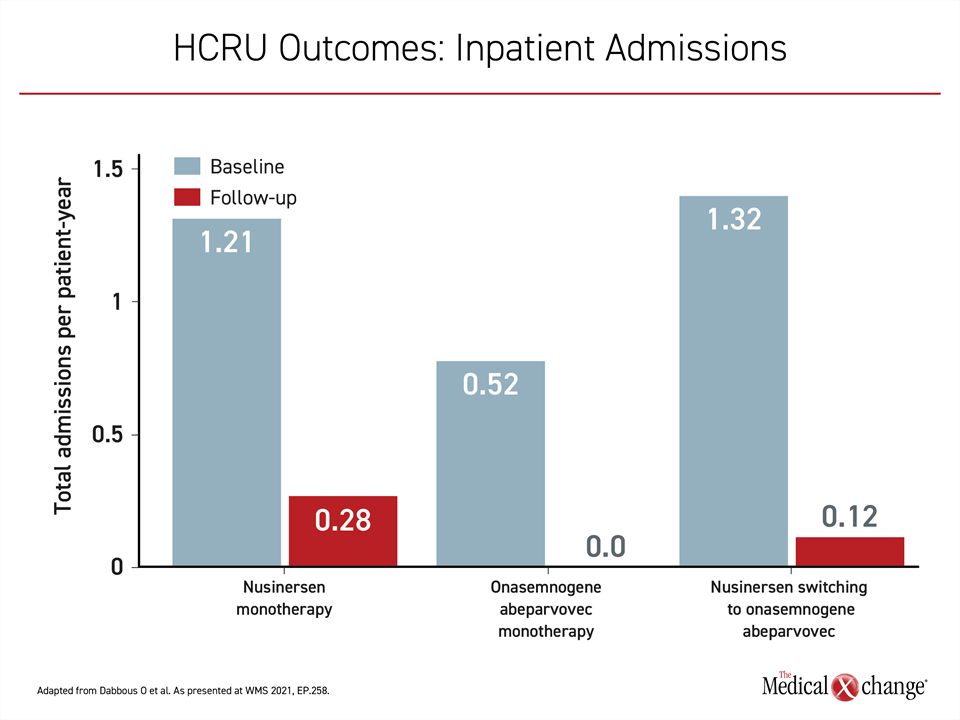
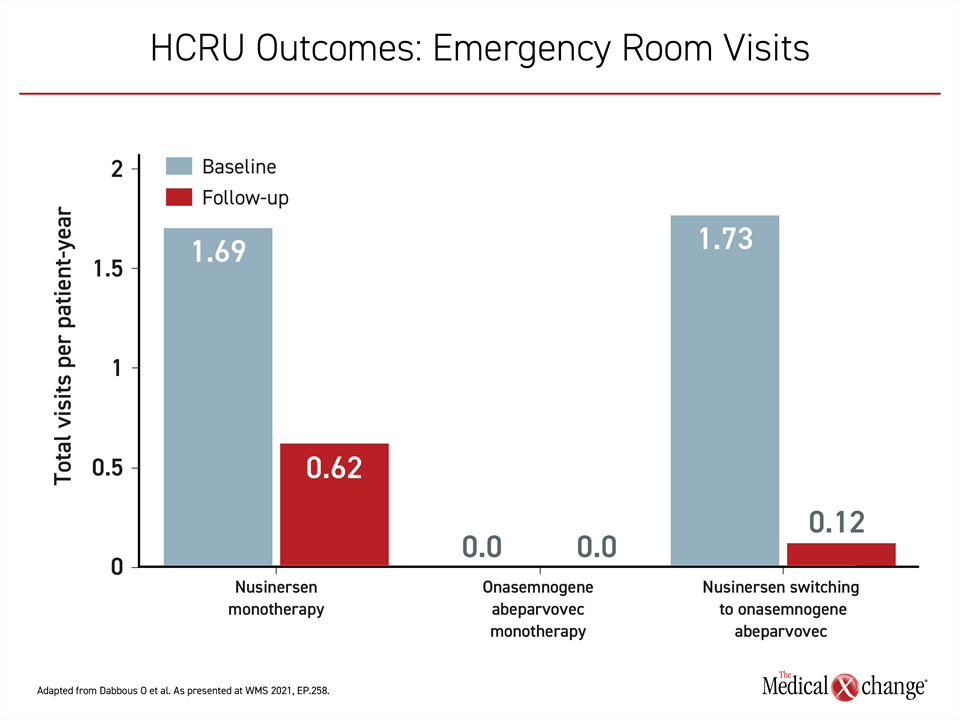
The HCRU study, based on a retrospective chart review, provided indirect support for the same conclusion. In this chart review, resource utilization was compared among those ≥6 months or older who received nusinersen monotherapy, onasemnogene abeparvovec monotherapy, or onasemnogene abeparvovec after prior nusinersen therapy. The primary outcomes were inpatient admission and emergency room (ER) visits before and after treatment administration.
For both of these outcomes, switching to onasemnogene abeparvovec was associated with lower resource utilization than nusinersen monotherapy. However, employing gene replacement therapy as a first-line strategy in older children was superior to either strategy. In those receiving gene replacement therapy upfront, there were no inpatient admissions or ER visits following treatment (Figure 2 and Figure 3).
Fewer Healthcare Consultations after Gene Replacement Therapy
Healthcare-related consultations for SMA declined following upfront onasemnogene abeparvovec (3.79 vs. 4.95) or after switching to onasemnogene abeparvovec after treatment with nusinersen (4.14 vs. 7.83). In contrast, consultations nearly doubled after initiating therapy with nusinersen (6.93 vs. 3.64).
“Patients treated with onasemnogene abeparvovec monotherapy or those switching to onasemnogene abeparvovec after nusinersen had lower SMA-related HCRU,” reported Dr. Eric Q. Wu, Healthcare Economist and Researcher, Analysis Group, Boston, Massachusetts.
This is the first study to compare HCRU following initiation of different types of disease-modifying therapies for SMA, according to Dr. Wu. He explained that the 20 patients included in this chart review had not participated in any prior clinical study. About half had type 1 SMA. Most of the others had SMA type 2. Eight patients were treated with nusinersen monotherapy, four with onasemnogene abeparvovec therapy monotherapy and eight with onasemnogene abeparvovec therapy after being switched from nusinersen.
HCRU Reductions Even After Switch of Treatment
“Because of the significant disease burden, SMA incurs substantial HCRU,” said Dr. Wu, citing previously-published data suggesting that costs remain high in SMA type 1 patients even after treatment with nusinersen. While his analysis did not look at specific costs directly, he believes that the relative reduction in hospitalizations and ER visits following one-time initiation of gene replacement therapy is meaningful.
Milestones Achieved Faster After Newborn Screening
The pivotal trials with onasemnogene abeparvovec, such as STR1VE-US, STR1VE-EU, and SPR1NT, associate this therapy with achievement of major milestones in most children with SMA type 1 treated within the first few weeks of life. Some children who were enrolled in the earliest clinical studies are functioning normally five years after treatment. This has increased a call for newborn screening.
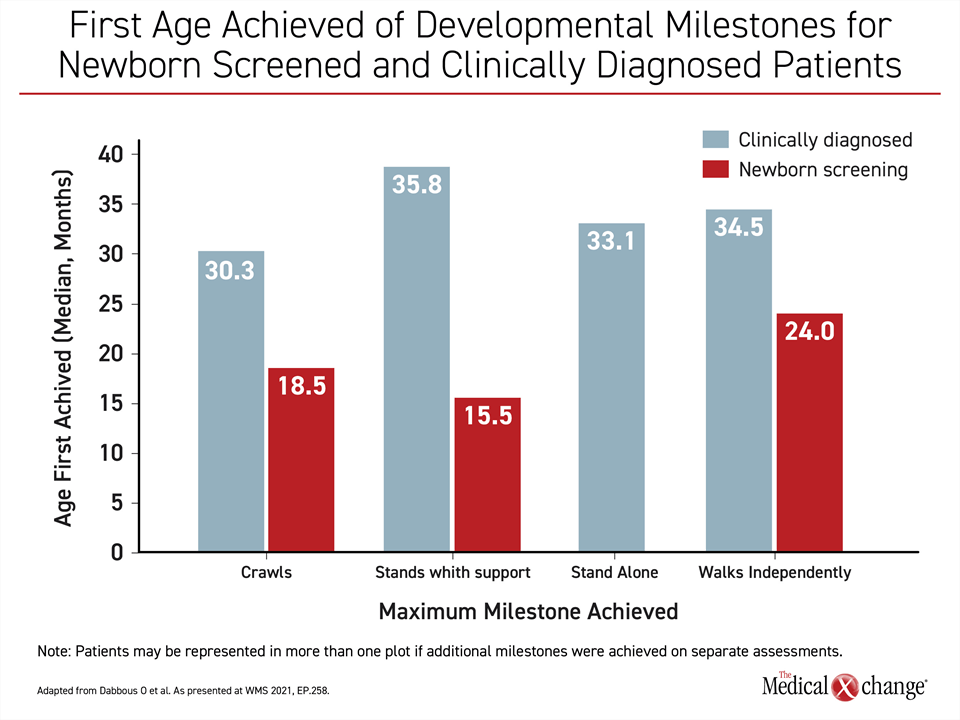
A second set of data drawn from the RESTORE registry and presented here provided data that supports this approach. In this analysis, 13 patients with SMA identified by newborn screening were compared to 42 patients who were clinically diagnosed. Outcomes at approximately two years after diagnosis strongly favored screening, most likely as a product of reducing the neurons permanently lost by earlier initiation of an effective treatment.
“Children identified with newborn screening generally achieved motor milestones at earlier ages than those clinically diagnosed, and a greater percentage identified by newborn screening achieving a ≥4 point increase in CHOP INTEND score during the period of observation,” reported Dr. Servais, who presented these data.
For clinically-diagnosed patients compared with those diagnosed through newborn screening, the mean age at diagnosis was 3.2 vs. 0.8 months (P<0.0001), and the age at first treatment was 4.9 vs. 1.7 months (P<0.0001), respectively. Although the time from diagnosis to first treatment (1.2 vs. 1.3 months: P=0.08) did not differ significantly, those diagnosed through newborn screening were less likely to receive more than one disease-modifying treatment (53.9% vs. 90.4%; P=0.012). Prompt initial treatment with the most effective therapy might explain the advantage of newborn screening across multiple milestones (Figure 4).
“The median age that newborn screening patients first achieved the Bayley-III milestone of independent sitting for ≥30 seconds was 12 months with those diagnosed through newborn screening versus 23.5 months for those who were clinically diagnosed,” Dr. Servais reported.
With the advent of more effective therapies for SMA, particularly in preventing fatal outcomes in children with SMA type 1, screening programs have been initiated in several centers, according to Dr. Servais, but he suggested there is increasing data, including these new data drawn from the RESTORE registry, that support uniform screening programs.
“Our study highlights the need to understand and overcome barriers to newborn screening implementation as well as to identify the reasons for delays to treatment once the diagnosis is made,” Dr. Servais said.
Conclusion
Over the past several years, the rapid progress in understanding and treating the underlying pathophysiology of SMA has provided an opportunity to save lives and reduce disability. Early administration of a single dose of gene replacement therapy permits normal motor development in a substantial proportion of SMA patients, including those with otherwise fatal type 1 disease. New data shows that gene replacement therapy also offers major benefits even in symptomatic, older children treated after 6 months of age. However, pre-symptomatic initiation of the most effective treatment provides the best outcomes, highlighting the importance of newborn screening. In at least some patients who receive gene replacement therapy early in life, normal motor function and development has been achieved in children followed more than 5 years.