Neurology
Progressive MS: Expert Review and Commentary from Published Literature
From Pathophysiology to Therapeutic Relevance in Progressive MS
V. Wee Yong, PhD, FCAHS, FRSC
Hotchkiss Brain Institute and the Department of Clinical Neurosciences
Cumming School of Medicine
University of Calgary
Calgary, Alberta
A more detailed understanding of the neuropathology of progressive multiple sclerosis (PMS) is producing new potential targets and strategies for disease control. PMS, like relapsing-remitting multiple sclerosis (RRMS), is driven by global brain atrophy, slowly expanding lesions, and inflammation, but dominant characteristics differ, including a more prominent role for activation of microglia, oxidative stress, and involvement of T and B lymphocytes compartmentalized beyond the blood-brain barrier (BBB). The relative role of therapies on these features and the degree of penetration into the central nervous system (CNS) parenchyma to attenuate inflammatory activity and other causes of neurodegeneration is likely to be essential for targeting PMS. These differences might explain why some existing therapies, such as siponimod, exhibit activity against PMS, while other therapies with an anti-inflammatory effect, such as teriflunomide, may not. The current understanding of PMS neuropathology suggests a greater opportunity for disease modification if treatment is started early in the course of injury to the CNS. It is also providing a framework for predicting which of the therapies in clinical development offer the greatest potential for control of PMS.
Background
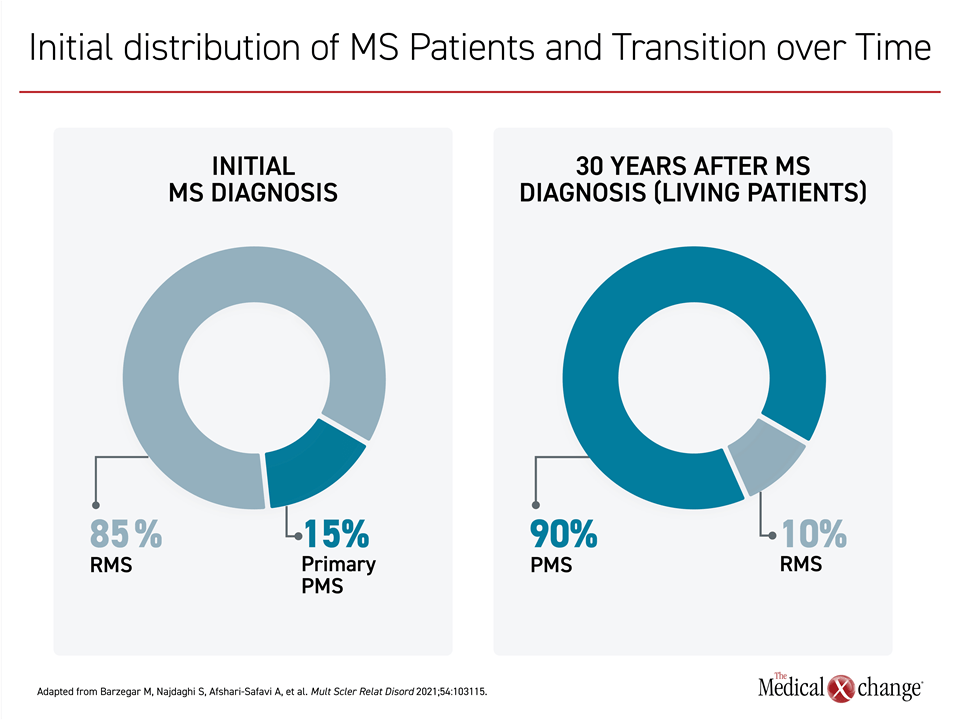
Three major phenotypes of multiple sclerosis (MS) are generally recognized, with RRMS representing the initial diagnosis in about 85% of patients.1 Natural history studies indicate that the majority of patients with RRMS transition to the secondary progressive (SPMS) phenotype within 20 years; however, this is variable as a small subset of patients never makes the transition, while it has occurred within a year in others.2 Approximately 15% percent of MS patients at diagnosis have progressive disease, called primary PMS (PPMS)1 (Figure 1). There are no universally accepted clinical, imaging, or biomarker criteria to define primary or secondary PMS, but an accumulation of sustained functional loss with few, diminishing, or no remitting symptoms provides a PMS diagnosis whether at disease onset or at the end of the RRMS phase.
For an exclusive interview with Dr. V. Wee Yong on the impact to clinical practice, click here
Due to differences in clinical course of PMS relatively to RRMS, the hypothesis that these might represent distinct disease entities has persisted for decades.3,4 However, it has become clear that the neuropathological features within the CNS differ in extent, rather than absolute or binary, when the progressive phenotypes are compared to the RR presentation.4-6 The inability of most therapies approved for RRMS to achieve meaningful benefit in PMS might be related to these differences. This provides a basis for postulating that viable targets for disease modification of RRMS and PMS phenotypes will differ for the goal of modifying pathology that drives symptoms and progression.
For example, even though lymphocyte infiltration is a prominent feature of RRMS, it tends to be limited to perivascular aggregation in this phenotype.7,8 This vascular inflammation plays a role in disrupting the BBB, which might play a role in the transition to PMS.9 Lymphocytes tend to be diffusely distributed within the CNS parenchyma in PMS, and the BBB is relatively intact in these phenotypes.6,8 The result is the compartmentalization of lymphocytes within the CNS parenchyma8 and sequestering these cells from therapies that do not cross the BBB. Moreover, the predominant microglia/macrophage-mediated inflammation within the CNS parenchyma of PMS has often been contrasted to the substantial lymphocyte infiltration that characterizes RRMS.7
There are other examples of quantitative rather than absolute differences in PMS relative to RRMS neuropathology,6 but the compartmentalization of lymphocytes is a good example as to why therapies for RRMS are not necessarily effective for PMS.
Current Treatments: Activity in PMS
After an extended period in which beta interferons and glatiramer acetate were the only approved therapies for RRMS, nearly 20 additional disease-modifying therapies (DMTs) have been made available in Canada over the past 15 years. The proliferation of therapies for RRMS has established the value of numerous targets, such as B-cells and transcription factors, to attenuate damage to the CNS during the period of relapsing-remitting disease. In contrast, there are so far only two approved therapies in Canada for PMS phenotypes. Both were approved relatively recently, but progress in therapeutics is paralleling advances in characterizing the neuropathology of PMS. Both are providing a framework for understanding how the disease course can be modified while offering clues toward development of a new generation of drugs.
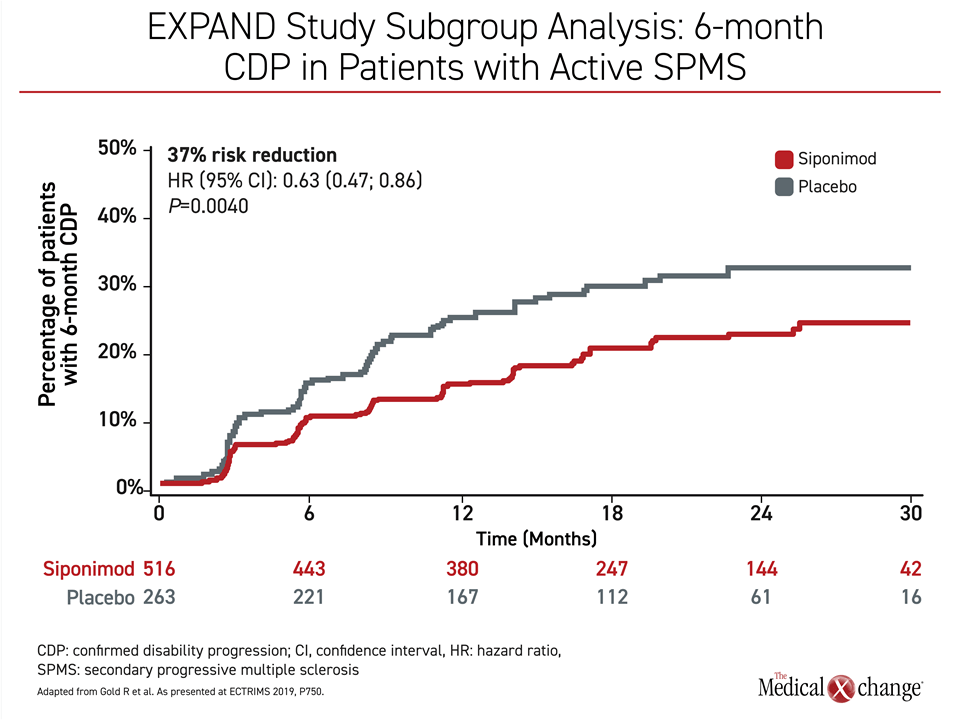
In Canada, one drug, the sphingosine 1-phosphate receptor (S1PR) modulator siponimod, is a licensed agent for the treatment of SPMS. The anti-CD20 monoclonal antibody ocrelizumab is approved for use in PPMS. The approval of siponimod was granted on the basis of the multicenter phase 3 EXPAND trial that associated this therapy with a 21% (P=0.013) reduction in confirmed disability progression relative to placebo at 12 weeks10. In an EXPAND study subgroup analysis, the risk of 6-month confirmed disability progression was reduced by 37% (P=0.004) in patients with active SPMS (Figure 2)11. As a modulator of the S1PR, siponimod regulates lymphocyte recirculation but it has other characteristics observed clinically or in experimental models that are likely relevant to activity in PMS, including its ability to cross the BBB,12 attenuate microgliosis and astrogliosis,13 and reduce oxidative stress and mitochondrial injury.14
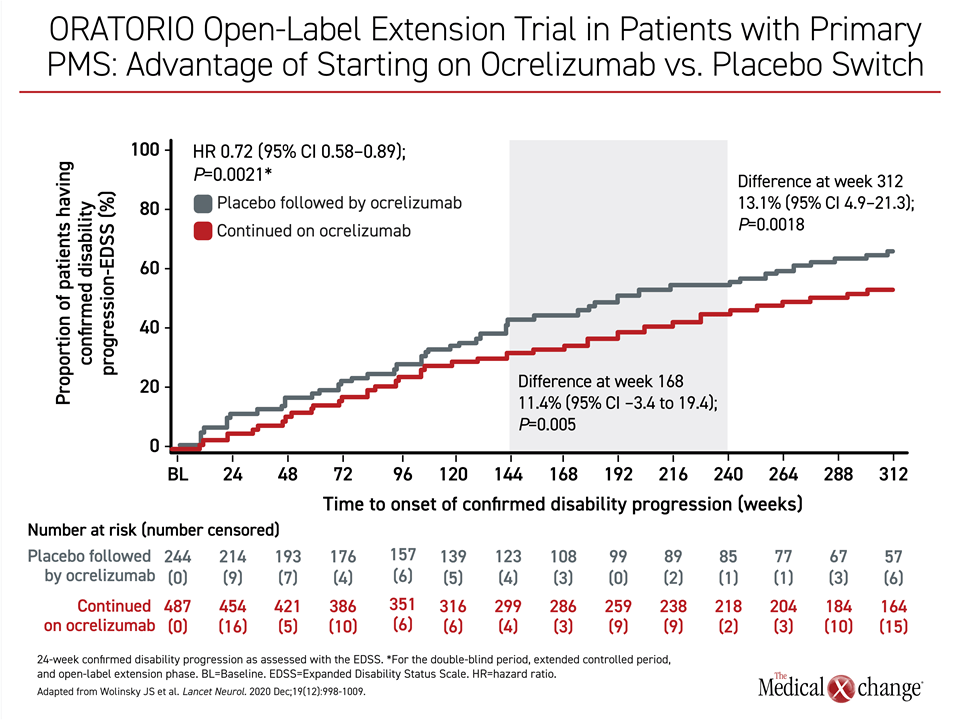
Ocrelizumab was granted a conditional approval for primary PMS on the basis of the multicenter phase 3 ORATORIO trial that described a 25% reduction in confirmed disability progression relative to placebo at 12 weeks.15 After more than 5 years of follow-up in an open-label extension, the proportion of patients with 6-month confirmed disability progression was reduced 13% (P=0.0018)16 (Figure 3). Despite the clinical benefit observed in the ORATORIO trial, the large molecular size of monoclonal antibodies argues against substantial BBB penetration,17 a limitation that raises concern about a sustained effect.
Neuropathology of PMS: Six Criteria to Impact Strategies for Therapy
In predicting therapies likely to inhibit disease activity in PMS, the relative ability of therapies to cross the BBB is reasonably considered an essential feature of drugs with a disease-modifying potential, but there are others. In a recently-published review of PMS neuropathology, a total of 6 criteria were identified as likely predictors of benefit. None are proven features of effective therapy, but, like the ability to penetrate the BBB, they are reasonable based on the characterization of PMS neuropathology.6
In defining how PMS differs from RRMS, accumulation of disability is reflected by the accelerated disease process on imaging and histochemical analysis. Relative to RRMS, PMS imposes a greater degree of neuroaxonal loss per lesion volume.18 The plaque-like slowly expanding lesions (SELs) typical of PMS and less prominent in RRMS, show radial expansion in the white matter, contributing to atrophy that is likely a contributor to neurologic dysfunction. Importantly, there has been progress in understanding the drivers of SELs, several of which appear to be targetable.
Specifically, within the compartmentalized neurodegeneration that characterizes PMS, there is evidence that oxidative stress, mitochondrial dysfunction, and iron toxicity, which are less well described in RRMS, are all important and contributing factors in PMS phenotypes.6 Although most of these factors can be detected at the earliest stages of disease, they assume a greater role in PMS, when microglia, in particular, switches from a reparative role that includes phagocytic removal of myelin debris, and begins to contribute to participate in neurodegeneration.19
Based on these features of disease, attractive characteristics of agents designed for PMS would, in addition to the ability to cross the BBB, inhibit lymphocyte activity, particularly the cytotoxicity of B cells in the CNS,20 attenuate microglial reactivity,21,22 reduce oxidative stress through antioxidant activity,23 promote remyelination,24 and exert neuroprotection.6
Some or all of these targets might also be relevant to disease-modifying effects in RRMS, but there is a basis to argue that most or all are required for disease-modifying effects in PMS. The greater isolation of inflammatory processes in the CNS parenchyma, the evidence of more pronounced pathogenicity exerted by oxidative stress and iron toxicity, the higher microglial reactivity, and the greater loss of myelin, indicate that disease progression is driven by multiple processes.6 On a theoretical basis, favorable effects on these targets predict the attenuation of disease processes, but they also correspond with features of drugs that have shown activity in PMS.
Most of the agents currently licensed for RRMS have at least one of these characteristics, but fewer have activities on 3 or more. Exceptions include cladribine, which is likely to cross the BBB, depletes lymphocytes in the CNS and inhibits microglia reactivity, but it is not known to have any antioxidant effect. Dimethyl fumarate, which also crosses the BBB, has also been associated with lymphocyte depletion and has an antioxidant effect, but is not known to inhibit microglial reactivity (Table 1).
Except for siponimod, which meets all six criteria, the only currently-approved agent to meet more than three criteria is fingolimod, which meets 5. Like siponimod, fingolimod also regulates S1P1, but has less selectivity, blocking the activity of S1P3, S1P4, and S1P5 as well. Siponimod also differs for its pharmacokinetic characteristics in the CNS, and it is not a prodrug. Fingolimod has not been clearly associated with remyelination directly, although studies have been conflicting. In addition, fingolimod, unlike siponimod, has not been associated with a disease-modifying effect for PMS in a major trial. In a phase 3 study that enrolled patients with primary PMS, fingolimod was not associated with significant protection against accumulation of disability.25
Targets of Disease Modification in PMS
Early in its course, MS is a disease of upregulated inflammatory factors, but PPMS and the transition from RRMS to SPMS appears to involve a far more complex interplay of factors that lead to CNS injury. Inhibition of conventional components of an inflammatory response, such as T and B cells, appears to be important but insufficient when neuronal loss advances in PMS. As suggested by the activation of microglia and macrophages, by neuronal damage exacerbated with oxidative stress and iron deposition, and by increasing mitochondrial dysfunction, it is likely that effective therapy will depend on action against multiple targets.
Many novel therapies are being pursued. For example, the antioxidant alpha-lipoic acid (ALA), an endogenous chemical that readily crosses the BBB, has already been tested in a controlled trial in SPMS, demonstrating protection against loss of brain volume and a trend for protection against disability.26 Inhibitors of Bruton’s tyrosine kinase (BTK), which appear to mediate microglial activity, have an antioxidant effect while inhibiting the activity of B cells. While several trials are ongoing with BTK inhibitors in RRMS, phase 3 trials in PMS are now enrolling patients, according to clinicaltrials.gov, which lists studies with tolebrutinib, remibrutinib and fenebrutinib. Similarly, the phosphodiesterase inhibitor ibudilast, which also mediates microglial activity, has shown more encouraging disease-modifying effects in early trials conducted in patients with primary PMS than it has in SPMS, suggesting that key targets in these two PMS phenotypes may differ.27
The signals of benefit from these experimental therapies are encouraging. The first therapy to be approved for PMS in Canada was ocrelizumab in 2018. Siponimod, the second, was approved in 2020 for SPMS. These therapies establish a foothold for considering new strategies for addressing the pathogenic mechanisms of PMS. It is reasonable to speculate that the greatest benefits from disease modification will be achieved early before irreversible CNS injury. If the underlying causes of PMS can be addressed, early treatment might include preventing the transition from RRMS to SPMS or at least modifying the neural substrates of disease progression. With evidence that some of the pathogenic features of SPMS are present during RRMS, the progress in understanding SPMS might lead to new opportunities to preserve neurologic function.
Summary
In an examination of the neuropathology of PMS, six criteria have been identified for agents with the greatest likelihood to inhibit disease progression. By targeting most or all of these criteria, which include penetration of the BBB, inhibition of lymphocyte activity, inhibition of microglial reactivity, reduction of oxidative stress, promotion of remyelination, and neuroprotection, the goal is to restore or at least preserve CNS function, preventing disability. The efficacy of siponimod, which meets these criteria, indirectly supports the value of these targets, while providing a direction for identifying additional therapies with disease-modifying effects on PMS.
References
1. Barzegar M, Najdaghi S, Afshari-Safavi A, et al. Mult Scler Relat Disord 2021;54:103115. DOI: 10.1016/j.msard.2021.103115
2. Cree BAC, Arnold DL, Chataway J, et al. Secondary Progressive Multiple Sclerosis: New Insights. Neurology 2021;97(8):378-388. DOI: 10.1212/WNL.0000000000012323.
3. Olerup O, Hillert J, Fredrikson S, et al. Primarily chronic progressive and relapsing/remitting multiple sclerosis: two immunogenetically distinct disease entities. Proc Natl Acad Sci U S A 1989;86(18):7113-7. DOI: 10.1073/pnas.86.18.7113.
4. Antel J, Antel S, Caramanos Z, Arnold DL, Kuhlmann T. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol 2012;123(5):627-38. DOI: 10.1007/s00401-012-0953-0.
5. Lassmann H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front Immunol 2018;9:3116. DOI: 10.3389/fimmu.2018.03116.
6. Yong HYF, Yong VW. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat Rev Neurol 2022;18(1):40-55. DOI: 10.1038/s41582-021-00581-x.
7. Weiner HL. A shift from adaptive to innate immunity: a potential mechanism of disease progression in multiple sclerosis. J Neurol 2008;255 Suppl 1:3-11. DOI: 10.1007/s00415-008-1002-8.
8. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 2012;8(11):647-56. DOI: 10.1038/nrneurol.2012.168.
9. Ortiz GG, Pacheco-Moises FP, Macias-Islas MA, et al. Role of the blood-brain barrier in multiple sclerosis. Arch Med Res 2014;45(8):687-97. DOI: 10.1016/j.arcmed.2014.11.013.
10. Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet 2018;391(10127):1263-1273. DOI: 10.1016/S0140-6736(18)30475-6.
11. Gold R, Kappos L, Bar-Or A, et al. Efficacy of Siponimod in Secondary Progressive Multiple Sclerosis Patients With Active Disease: The EXPAND Study Subgroup Analysis. P750. 35th Congress of the European Committee for Treatment and Research in Multiple Sclerosis, September 2019..
12. Scott LJ. Siponimod: A Review in Secondary Progressive Multiple Sclerosis. CNS Drugs 2020;34(11):1191-1200. DOI: 10.1007/s40263-020-00771-z.
13. Gentile A, Musella A, Bullitta S, et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J Neuroinflammation 2016;13(1):207. DOI: 10.1186/s12974-016-0686-4.
14. Ward LA, Lee DS, Sharma A, et al. Siponimod therapy implicates Th17 cells in a preclinical model of subpial cortical injury. JCI Insight 2020;5(1). DOI: 10.1172/jci.insight.132522.
15. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med 2017;376(3):209-220. DOI: 10.1056/NEJMoa1606468.
16. Wolinsky JS, Arnold DL, Brochet B, et al. Long-term follow-up from the ORATORIO trial of ocrelizumab for primary progressive multiple sclerosis: a post-hoc analysis from the ongoing open-label extension of the randomised, placebo-controlled, phase 3 trial. Lancet Neurol 2020;19(12):998-1009. DOI: 10.1016/S1474-4422(20)30342-2.
17. Banks WA. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol 2009;9 Suppl 1:S3. DOI: 10.1186/1471-2377-9-S1-S3.
18. Matthews PM, Pioro E, Narayanan S, et al. Assessment of lesion pathology in multiple sclerosis using quantitative MRI morphometry and magnetic resonance spectroscopy. Brain 1996;119 ( Pt 3):715-22. DOI: 10.1093/brain/119.3.715.
19. Jackle K, Zeis T, Schaeren-Wiemers N, et al. Molecular signature of slowly expanding lesions in progressive multiple sclerosis. Brain 2020;143(7):2073-2088. DOI: 10.1093/brain/awaa158.
20. Lisak RP, Nedelkoska L, Benjamins JA, et al. B cells from patients with multiple sclerosis induce cell death via apoptosis in neurons in vitro. J Neuroimmunol 2017;309:88-99. DOI: 10.1016/j.jneuroim.2017.05.004.
21. Prineas JW, Kwon EE, Cho ES, et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol 2001;50(5):646-57. DOI: 10.1002/ana.1255.
22. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Bruck W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol 2013;125(4):595-608. DOI: 10.1007/s00401-013-1082-0.
23. Plemel JR, Liu WQ, Yong VW. Remyelination therapies: a new direction and challenge in multiple sclerosis. Nat Rev Drug Discov 2017;16(9):617-634. DOI: 10.1038/nrd.2017.115.
24. Fischer MT, Wimmer I, Hoftberger R, et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain 2013;136(Pt 6):1799-815. DOI: 10.1093/brain/awt110.
25. Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016;387(10023):1075-1084. DOI: 10.1016/S0140-6736(15)01314-8.
26. Spain R, Powers K, Murchison C, et al. Lipoic acid in secondary progressive MS: A randomized controlled pilot trial. Neurol Neuroimmunol Neuroinflamm 2017;4(5):e374. DOI: 10.1212/NXI.0000000000000374.
27. Fox RJ, Coffey CS, Conwit R, et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N Engl J Med 2018;379(9):846-855. DOI: 10.1056/NEJMoa1803583.