Neurology
American Academy of Neurology (AAN) Annual Meeting 2022
Gene Replacement Therapy Found to be Safe and Effective in Older Patients with Spinal Muscular Atrophy
Seattle – In its most common and severe form, the genetic neuromuscular disease spinal muscular atrophy (SMA) leads to symptoms within months of birth. When treated with a recently released gene replacement therapy, most children have age-appropriate early development when treated prior to the onset of symptoms. Although the value of gene therapy in symptomatic children 6 months or older has been less well studied, a prospective registry has generated data to suggest that children treated up to 24 months of age benefit from gene replacement therapy. Whether ≥6 months or ≥12 months, both groups achieved neuromuscular functional gains. The registry data show good tolerability and efficacy even though older, heavier children receive higher doses of the weight-based gene replacement therapy.
The gene replacement therapy onasemnogene abeparvovec was approved in Canada at the end of 2020. Unlike nusinersen and risdiplam, the other two licensed therapies for SMA, gene replacement therapy restores production of the deficient SMN1 protein after a single injection, eliminating the need for maintenance doses. There have been limited data with this drug when initiated after symptom onset; this is changing as a result of the multinational observational RESTORE registry (clinicaltrials.gov; NCT04174157). In this registry, which is enrolling children with SMA irrespective of treatment, benefit following symptom onset has been documented.
For an exclusive interview with Dr. Nicolas Chrestian on the impact to clinical practice, click here
RESTORE Registry Provides Real-world Data in Older Children
“Onasemnogene abeparvovec is an effective therapeutic choice in older and heavier children, and this includes children treated previously with another therapy,” reported Dr. Richard S. Finkel, Director of the Experimental Neuroscience Program, St. Jude’s Children’s Hospital, Memphis, Tennessee.
“[We have] an effective therapeutic choice in older and heavier children, and this includes children treated previously with another therapy.”
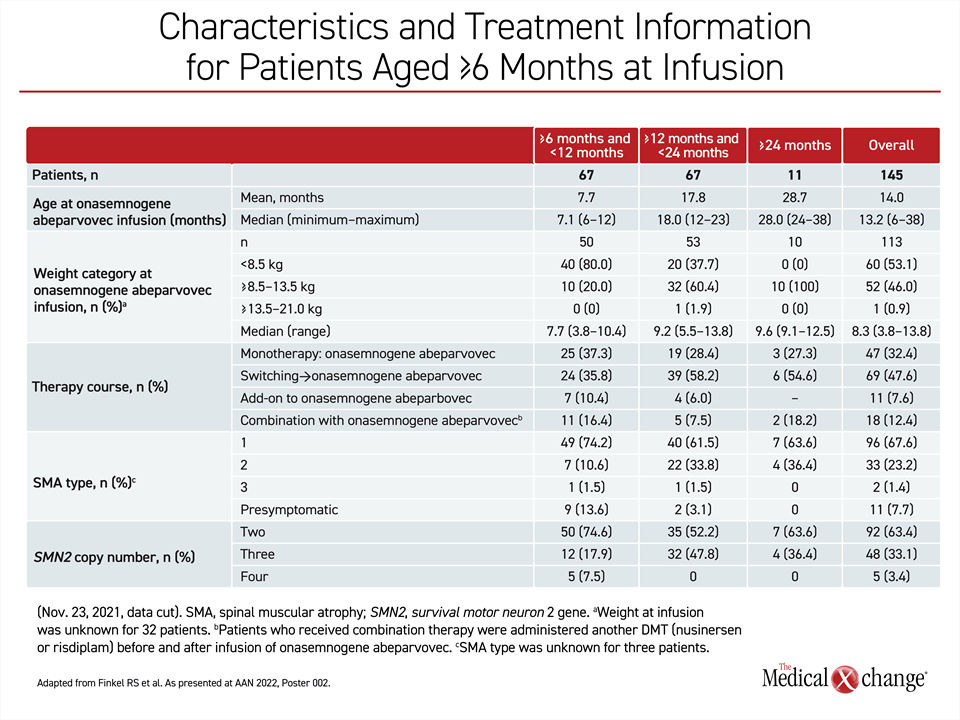
SMA is relatively rare, but the RESTORE registry has already enrolled more than 300 patients. This includes an analysis of 145 older children with SMA who were first treated with onasemnogene abeparvovec at 6 months or older in one of four therapy courses, including as a monotherapy, after a switch from another disease-modifying treatment (DMT), another DMT added to the gene replacement therapy, or in combination with another DMT. Of these, 67 children were ≥6 to <12 months (group 1) and another 67 were ≥12 to <24 months (group 2). Another age group included eleven children who were first treated at ≥24 months (group 3), which is out of labelling but reflective of the realities of a real-world registry.
Motor Function Assessment in Older Children
Several measures of response and safety were evaluated, but CHOP INTEND (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders) score served as the primary efficacy outcome of the most recent analysis. CHOP INTEND uses a 0- to 64-point rating for neuromuscular function with a change of ≥4 points considered to be clinically meaningful. This tool was developed specifically for monitoring SMA.
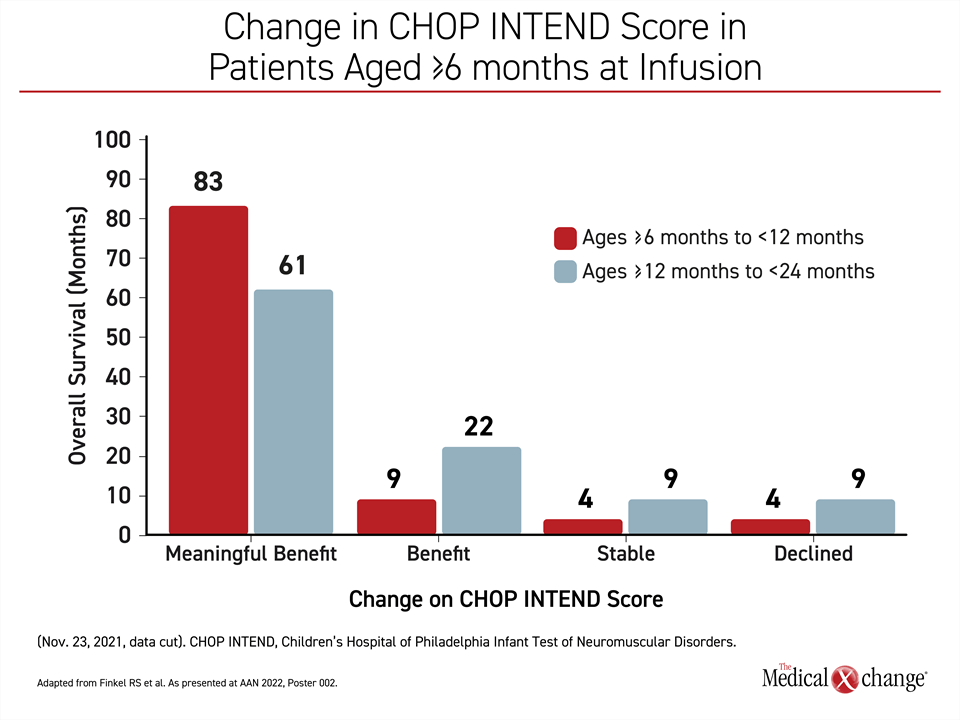
On the basis of ≥4-point improvement, 19 (83%) of the 23 patients in group 1 who are so far evaluable (≥2 CHOP INTEND assessments more than 2 months apart) met the criterion for a meaningful benefit. Two of the others (9%) had an increase in score below 4 points. One had a stable score, and one had a decline.
Similarly, 14 (61%) of the 23 evaluable older patients in group 2 met the criterion for a meaningful benefit. Of the remaining patients, 5 (22%) had an increase in score below 4 points, 2 patients had a stable score, and two had a decline (Figure 1).
None of the group 3 patients have been evaluable, but these, along with the 44 children in group 1 and another 44 children in group 2 will continue to be monitored in the RESTORE registry. This registry has a planned enrollment of more than 500 children around the globe. The planned follow up is 15 years.
Many SMA Types Are Fatal Without Therapy
SMA is caused by a missing or deleted SMN1 gene. The type 1 phenotype is the most severe and the most common, but type 2 phenotype patients, like type 1, typically die in early childhood without treatment. The number of SMN2 gene copies is a factor in determining severity. Like the SMN1 gene, SMN2 can also produce the SMN1 protein, but it does so in smaller quantities. Most type 1 patients have just two SMN2 gene copies. Relative to the type 2 phenotype, type 1 patients produce smaller amounts of the SMN1 protein and tend to have earlier onset of symptoms and a more rapidly progressive disease.
When all 67 patients enrolled in the RESTORE registry at ≥6 to <12 months of age are compared to the 67 patients enrolled at ≥12 to <24 months of age, more of the younger patients had the type 1 phenotype (74% vs. 62%). They were less likely to have the type 2 phenotype (11% vs. 34%). For most of the others in both groups, patients were enrolled when they were pre-symptomatic, so phenotype has not yet been determined.
Differences in the timing and severity of symptoms might explain patterns of treatment in the RESTORE registry. Younger patients were more likely to receive onasemnogene abeparvovec as a monotherapy (37% vs. 28%) and less likely to be switched to onasemnogene abeparvovec after receiving another DMT (36% vs. 58%). More of the younger patients were also more likely to receive onasemnogene abeparvovec in combination with another DMT or as an add-on to a prior therapy (27% vs. 14%) (Table 1).
Gene Replacement Therapy Beneficial Even in Oldest Group Treated
The higher proportion of younger patients who achieved a meaningful improvement in the CHOP INTEND score is consistent with the advantages of increasing functional SMN1 protein levels early in the course of disease, but it is important to emphasize that most patients in both age groups benefited. Dr. Finkel also emphasized that the greater than 80% response rate in the younger patients was achieved even though the majority had the most severe phenotype. SMN2 gene copy number was not a predictor of response in these data.
“When you compare those with 2 versus 3 [SMN2] copies, we are showing pretty equal improvement. Copy number does not appear to be a significant stratifying factor.”
“When you compare those with 2 versus 3 [SMN2] copies, we are showing pretty equal improvement. Copy number does not appear to be a significant stratifying factor [for response to gene replacement therapy],” he said.
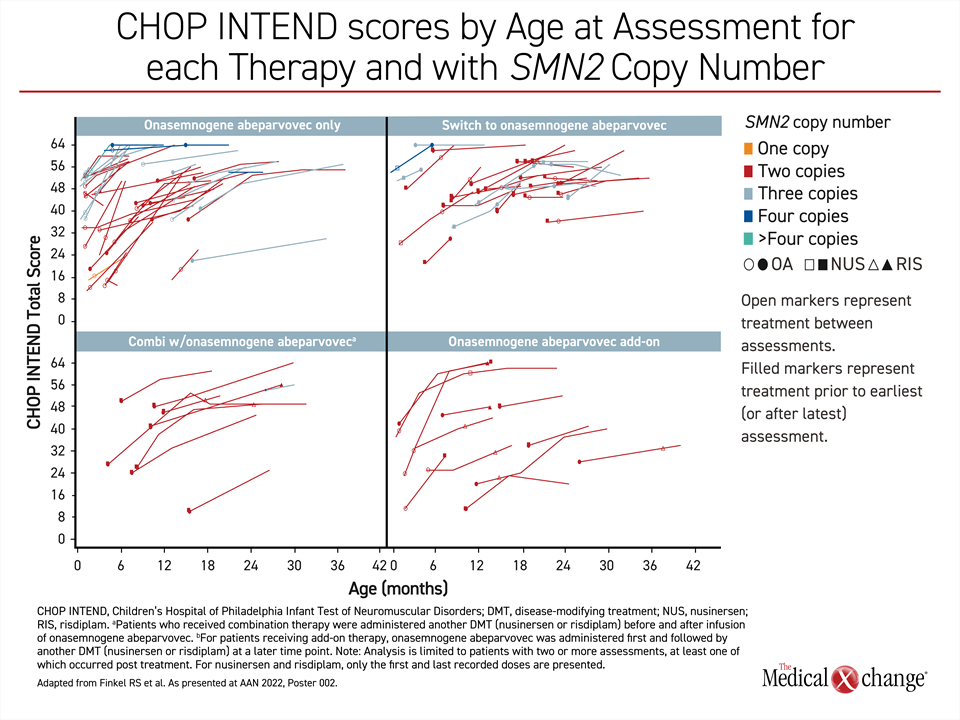
Overall, there was about a 1-point-per-month improvement in CHOP INTEND score across groups in an observation period that is out to about 10 months. The response so far is not just independent of copy number but also independent of stratification— whether patients received onasemnogene abeparvovec as monotherapy, after another DMT, in combination with another DMT, or as an add-on to a previous therapy.
“In looking at the curves, I think it can be appreciated that almost all the older patients were generally improving.”
“Almost every curve shows an upward trajectory,” Dr. Finkel said. Although he acknowledged that the gradient or improvement was less steep in older relative to younger patients, “I think it can be appreciated that almost all patients were generally improving” (Figure 2).
Safety in Older SMA Patients Reassuring
Along with effectiveness, the safety of onasemnogene abeparvovec among older children has also been a major focus of the RESTORE registry analysis. This is due not only to the potential for different safety issues in older children, but also because of weight-based dosing. The standard dosage of this agent is 1.1 x 1014 vector genomes per kilogram of body weight delivered in a single one-hour infusion. As children gain weight with age, they receive much higher vector exposures. However, the safety data from the registry were reassuring.
“The adverse event profile in patients treated at ≥6 months is consistent with the overall adverse event profile previously reported for onasemnogene abeparvovec,” Dr. Finkel said. “There was no pattern of greater risk of adverse events or adverse event severity when compared by age groups or by patient weight.”
“The adverse event profile in patients treated at ≥6 months is consistent with the overall adverse event profile previously reported.”
As reported in the prospective trials, elevated liver enzymes, reduced platelets and elevated troponin were observed in the RESTORE registry. Most hepatotoxicity in this series was asymptomatic, although two children developed acute liver failure. One was 6 months of age and the other 21 months. There were no cases of symptomatic thrombocytopenia or troponin elevations leading to clinical consequences. Four cases of thrombotic microangiopathy following onasemnogene abeparvovec were reported previously, but there have been no additional cases. In this series, the rates of any serious adverse event (33.3% vs. 26.5%) and grade ≥3 adverse events (36.4% vs. 26.5%) were lower in the older relative to the younger age group.
Early Diagnosis and Treatment of SMA Remains Urgent
In the real-world data produced by the RESTORE registry, onasemnogene abeparvovec “is an effective therapeutic choice in older patients regardless of prior DMT,” Dr. Finkel contended. Still, these data do not lessen the importance of diagnosing SMA early and starting therapy as soon as possible after birth, and preferably before symptoms.
“In the United States, 39 of 50 states now have newborn screening panels that include SMA,” Dr. Finkel said. This is important, because the phase 3 data with onasemnogene abeparvovec support age-appropriate early development in most children treated pre-symptomatically. The benefit of gene replacement therapy has created urgency for early treatment in type 1 patients with just 2 SMN2 copies. Failure to develop gross motor skills as a result of SMN1 protein deficiency might not be readily apparent to non-specialists until several months of age, but Dr. Finkel said that babies with SMA often do not seem to be developing normally within weeks of birth.
All Patients Treated Early Showed Improvement
At the AAN, there was a review of the previously presented SPR1NT phase 3 study and the results were impressive. In a cohort of SMA patients with 2 SMN2 copies (there is a separate analysis of SMA patients with 3 SMN2 copies), all 14 children received onasemnogene abeparvovec within 34 days of birth and then were followed for 18 months. The earliest treatment was 8 days after birth. Of the 14, all are sitting independently. Eleven of these did so within the expected age-appropriate developmental window.
By 18 months, when a substantial proportion of untreated children with SMA would be expected to require feeding and perhaps respiratory support, 11 were standing independently and nine were walking independently. Five of those walking independently did so within the expected age-appropriate developmental window. All children remain alive and none require any form of feeding or ventilatory support.
The benefits of gene replacement therapy have been shown to extend to older children in the real-world RESTORE registry. However, more follow up is needed to gauge the value of this therapy relative to the intrathecal administration of nusinersen or daily oral administration of risdiplam for sustained preservation of milestones. It is likely that children who do not receive therapy in advance of symptoms might never attain lost milestones, but the gains in neuromuscular function even with delayed gene replacement therapy appear to be clinically meaningful.
Summary
Gene replacement therapy for SMA, a congenital disorder that leads to progressive loss of neuromuscular function, has been available in Canada for more than 1 year. This therapy was approved based on phase 3 trials largely conducted in asymptomatic children. These studies demonstrated that many children achieve age-appropriate developmental milestones after a single dose of this therapy. Data from the RESTORE registry now provide evidence that meaningful benefit can also be achieved in children treated even after SMA symptoms develop, including children older than 12 months. The higher dose of weight-based gene replacement therapy in older and heavier children appears to be well tolerated as it is in younger patients.