Neurology
hATTR-PN: Expert Review and Commentary
Expanding Options for Hereditary Transthyretin Amyloidosis with Polyneuropathy
Vera Bril, BSc, MD, FRCPC
Professor of Neurology, University of Toronto
Neurologist and Director, Neuromuscular Section, University Health Network and University of Toronto
Senior Scientist, Toronto General Hospital Research Institute
Toronto, Ontario
Geneviève Matte, MDCM, FRCPC
Neurologist, Centre Hospitalier de l’Université de Montréal (CHUM)
Assistant Clinical Professor, Department of Neurosciences, Université de Montréal
Director, ALS and Motor Neuron Disease Clinic, CHUM
Montreal, Quebec
Transthyretin (TTR) gene silencer therapies are changing the natural history of hereditary transthyretin amyloidosis with polyneuropathy (hATTR-PN). Administered by injection or infusion, these therapies inhibit production of the TTR protein, which, when produced by a mutated TTR gene, misfolds and aggregates in peripheral and autonomic nerves to impair their function. The same mechanism is relevant to other ATTR subtypes, which are defined by predominant clinical features, but like hATTR-PN, typically involve multiple systems. In the phase 3 NEURO-TTRansform trial, the newest of these therapies was associated with substantial reductions in serum TTR concentration, a reduction in neurological impairment, and improvement in quality of life. The potential of this and other TTR silencer therapies to extend survival is being monitored in ongoing follow-up.
Background
Amyloidosis is a term describing a heterogeneous array of acquired and genetic disorders in which fibrillar proteins aggregate in target tissues to produce dysfunction.1 Unlike immunoglobulin light chain amyloidosis, the most common form of this disorder,2 ATTR manifests as a consequence of aging or as an autosomal-dominant familial condition in which mutations in the TTR gene is the key pathogenic mechanism.3 The most common subtypes are hATTR-polyneuropathy (PN), which involves amyloid fibril deposition in the autonomic and peripheral nerves,4 and ATTR-cardiomyopathy (CM), which is characterized by amyloid fibril deposition in the heart.5 Defined by their predominant clinical features, the progression and life-threatening dysfunction produced in patients with either subtype can be disseminated across organ systems.6
For an exclusive interview with Dr. Vera Bril on the impact to clinical practice, click here
Once better known as familial amyloid polyneuropathy, hATTR-PN, which was first described in 1952,7 has been considered rare or uncommon outside of endemic countries, such as Portugal, where the first cases were isolated.7 In a 2016 survey, the prevalence of hATTR-PN in Portugal was an estimated 23 cases per 100,000 adult inhabitants.8 It is unclear if this is representative of worldwide distribution, but cases have now been identified globally.9
The considerable heterogeneity in the presentation of hATTR-PN and ATTR-CM, including the symptoms and their severity, are primarily explained by differences in the underlying TTR mutations. In a recent review, more than 150 TTR mutations were counted in the Hereditary Amyloidosis Database.4 For hATTR-PN, the first identified TTR mutation, Val30Met, is also the most common, accounting for about 70% of cases in areas with endemic disease.6 Most of the TTR gene abnormalities associated with ATTR are missense variants produced by point mutations.
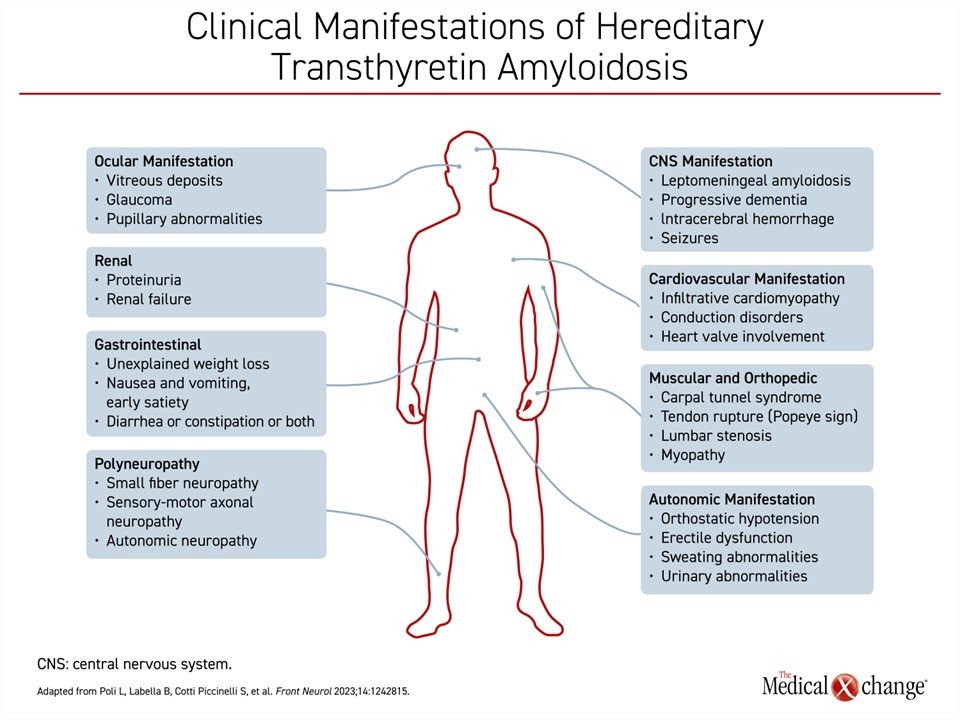
Beyond dysfunction produced in the peripheral or autonomic nervous system and the heart, ATTR can produce life-threatening dysfunction in a variety of other organ systems, such as the kidneys, the eyes, and the gastrointestinal tract (Figure 1). The risk is at least partly defined by underlying TTR mutation. Progressive renal dysfunction, for example, develops in about one-third of patients with the Val30Met mutation.10
Imaging, including ultrasound, computed tomography (CT) and magnetic resonance imaging (MRI), can be used to assess involvement in peripheral nerves and the heart, although symptoms and their severity provide a common method of evaluating the burden and extent of the disease.
Treatment
Considering the dominant hepatic expression of TTR, liver transplantation has been employed for decades to extend survival in patients with ATTR, but it was no longer listed among first-line therapies in Canadian ATTR guidelines published in 2022.11 Although liver transplant has been associated with improved survival,12 there are several issues, including continued cardiac amyloid deposition after transplant,13 that render this treatment inferior to the pharmacological therapies that are now available.
TTR stabilizers were introduced more than 15 years ago, but these have been used primarily in the treatment of ATTR-CM. By preventing degradation of the TTR protein, both of the most commonly used TTR stabilizers, diflunisal and tafamidis, inhibit a key step in amyloid deposition.14,15 Despite positive studies,15,16 diflunisal has never received regulatory approval for ATTR-CM. Tafamidis does have an indication for ATTR-CM in Canada based on a phase 3 trial,17 but its use is widely restricted to specialists. Neither has an indication in Canada for hATTR-PN.
TTR gene silencer therapies were the first treatments approved in Canada with a specific indication for hATTR-PN. Inotersen, a first-generation antisense oligonucleotide (ASO) came first in 2018, and patisiran, a small interfering RNA (siRNA), followed. The mechanisms differ. Inotersen is delivered by weekly subcutaneous injections and binds to TTR messenger RNA (mRNA) to inhibit TTR protein synthesis.18 Patisiran, administered through intravenous infusion, is internalized by hepatocytes where it produces catalytic degradation of TTR mRNA, thereby reducing circulating TTR.19
In the inotersen registration trial (NEURO-TTR),20 172 patients with hATTR-PN were randomized in a 2:1 ratio to inotersen or placebo administered in weekly subcutaneous injections. The primary endpoint of the trial was the modified Neuropathy Impairment Score+7 (mNIS+7) score, where an increase indicated a decline in neurological function. The results showed a significantly lower increase in the mNIS+7 score for patients on the TTR silencer therapy compared to those on placebo (+5.8 vs. +25.5 points; P<0.001). There was also a significant improvement in QoL for inotersen relative to placebo. Side effects associated with inotersen included glomerulonephritis and thrombocytopenia, of which one case was fatal.
In the patisiran registration trial (APOLLO), 225 patients with hATTR-PN were randomized in a 2:1 ratio to patisiran or placebo infusion every 3 weeks.21 On the primary endpoint of change in the mNIS+7, the mean average change (-6.0 vs. +28.0 points; P<0.001) was highly favourable for patisiran. Secondary measures of gait speed and QoL were also significantly improved. The therapy was well tolerated, though injection site reactions were higher on patisiran (20% vs. 10%).
Following a phase 3 trial (HELIOS-A), vutrisiran,22 another siRNA therapeutic for hATTR-PN was granted Health Canada approval in 2023. In the registration trial, 164 patients were randomized in a 3:1 ratio to subcutaneous vutrisiran every 3 months or intravenous patisiran every 3 weeks. The placebo arm of APOLLO was used as an external control. The reduction in serum TTR concentration for vutrisiran was non-inferior to patisiran, and vutrisiran was relatively well tolerated.
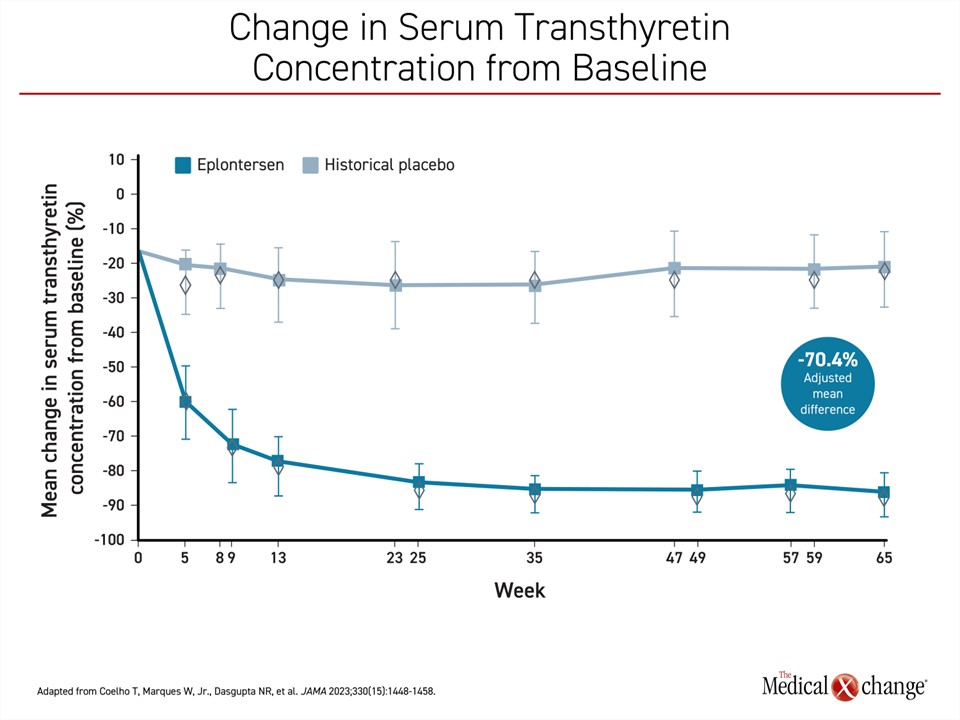
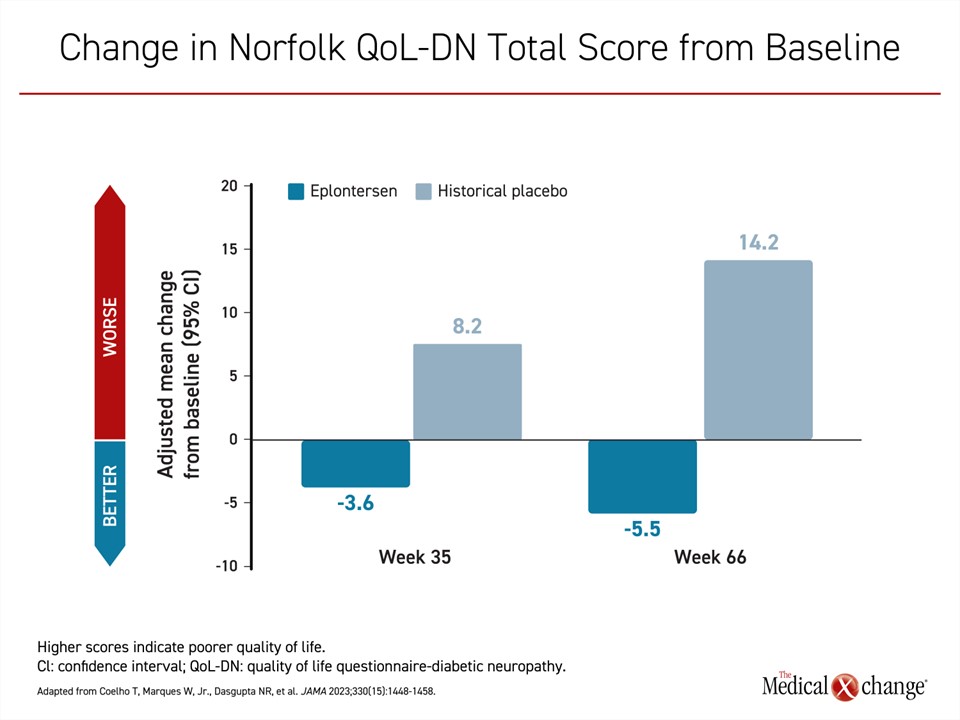
Eplontersen is the newest TTR silencer therapy to complete a phase 3 trial (NEURO-TTRansform).23 In this study, patients were randomized in a 6:1 ratio to open-label eplontersen or a reference treatment, inotersen. A historical placebo group from the previously published NEURO-TTR trial served as the comparator arm. In the published design of the trial, it was decided that randomization to a placebo arm was unethical given the availability of other therapies with established efficacy. The 24 patients in the inotersen reference group were transitioned to eplontersen at 34 weeks. The advantage of eplontersen relative to placebo on each of the three measures of the primary composite outcome at week 65 of the study were statistically highly significant. This included change in serum transthyretin concentration (-81.7% vs. -11.2%; P<0.001), mNIS+7 score (0.3 vs. 25.1; P<0.001) and change in the Norfolk Quality of Life Questionnaire-Diabetic Neuropathy (increasing scores equal worse quality of life) (-5.5 vs. 14.2; P<0.001) (Figures 2-4).
The rate of treatment discontinuations for an adverse event in the eplontersen and placebo groups was similar (4% vs. 3%). There were no cases of glomerulonephritis, and the rates of thrombocytopenia were the same in both groups at 2%. All cases of thrombocytopenia were mild and did not require dose changes or lead to bleeding events.
Eplontersen was not directly compared to the first-generation inotersen in this study, but differences in their mechanism of action might explain a low rate of adverse events and consistent efficacy for the newer agent. Like inotersen, eplontersen is an antisense oligonucleotide, but it employs novel ligand-conjugated technology. Conjugating N-acetyl galactosamine (GalNAc) to ASOs facilitates uptake by hepatocytes through asialoglycoprotein receptors. This is credited with improved targeting of the drug. An auto-injector facilitates self-administration and compared to inotersen, dosing intervals for eplontersen are much longer (every 4 weeks vs. weekly).
Challenges
The reduction in symptoms and improvement in quality of life over follow up that exceeds 1 year suggests that TTR gene silencer therapy alters the natural history of hATTR-PN. There are no direct comparisons between the TTR silencers, but the individual studies suggest they might not be comparable. All of the drugs have been effective relative to placebo but the delta in response as measured with mNIS+7 have varied, and the adverse event profiles differ.
Importantly, longer follow up is needed for all the available TTR silencer therapies to evaluate their potential to extend survival. A survival benefit might stem from relative inhibition of protein deposition across multiple organ systems not just from neuropathies. The first phase 3 study with a TTR silencer therapy in patients with ATTR-CM was recently completed with patisiran (APOLLO-B). It demonstrated preserved function at the end of 12 months.24 Results from a comparable trial with eplontersen (CARDIO-TTRansform) are expected soon.
Optimal benefit from TTR silencer therapy might depend on the initiation of treatment early in the disease, which, in turn, depends on earlier recognition of ATTR. Incremental gains, if any, from adding TTR stabilizers to TTR gene silencer therapy have not been explored.
Summary
Often associated with debilitating symptoms, hATTR-PN is a progressive and ultimately fatal disease if not controlled. An expanding list of TTR silencer therapies have been associated with an attenuation of disease progression and improvement in quality of life. The efficacy of eplontersen, the most recent of the TTR silencers for ATTR and the next generation in the ASO class, was established in the phase 3 NEURO-TTRansform trial. Self-administration by auto-injector with relatively long intervals between doses promises a convenient option. TTR silencer therapies have been a breakthrough for hATTR-PN and show promise for ATTR-CM. The extended follow-up studies will explore their ability to prevent damage across vulnerable organs and potentially extend survival.
References
- Gertz MA, Dispenzieri A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A Systematic Review. JAMA 2020;324(1):79-89. DOI: 10.1001/jama.2020.5493.
- Zanwar S, Gertz MA, Muchtar E. Immunoglobulin Light Chain Amyloidosis: Diagnosis and Risk Assessment. J Natl Compr Canc Netw 2023;21(1):83-90. DOI: 10.6004/jnccn.2022.7077.
- Gertz MA, Benson MD, Dyck PJ, et al. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J Am Coll Cardiol 2015;66(21):2451-2466. DOI: 10.1016/j.jacc.2015.09.075.
- Poli L, Labella B, Cotti Piccinelli S, et al. Hereditary transthyretin amyloidosis: a comprehensive review with a focus on peripheral neuropathy. Front Neurol 2023;14:1242815. DOI: 10.3389/fneur.2023.1242815.
- Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol 2019;73(22):2872-2891. DOI: 10.1016/j.jacc.2019.04.003.
- Luigetti M, Romano A, Di Paolantonio A, Bisogni G, Sabatelli M. Diagnosis and Treatment of Hereditary Transthyretin Amyloidosis (hATTR) Polyneuropathy: Current Perspectives on Improving Patient Care. Ther Clin Risk Manag 2020;16:109-123. DOI: 10.2147/TCRM.S219979.
- Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952;75(3):408-27. DOI: 10.1093/brain/75.3.408.
- Ines M, Coelho T, Conceicao I, Duarte-Ramos F, de Carvalho M, Costa J. Epidemiology of Transthyretin Familial Amyloid Polyneuropathy in Portugal: A Nationwide Study. Neuroepidemiology 2018;51(3-4):177-182. DOI: 10.1159/000490553.
- Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 2019;15(7):387-404. DOI: 10.1038/s41582-019-0210-4.
- Lobato L, Rocha A. Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol 2012;7(8):1337-46. DOI: 10.2215/CJN.08720811.
- Alcantara M, Mezei MM, Baker SK, Breiner A, Dhawan P, Fiander A. Canadian guidelines for Hereditary transthyretin amyloidosis polyneuropahty management. Can J Neurol Sci 2022;49:7-18.
- Yamashita T, Ando Y, Okamoto S, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology 2012;78(9):637-43. DOI: 10.1212/WNL.0b013e318248df18.
- Yazaki M, Mitsuhashi S, Tokuda T, et al. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am J Transplant 2007;7(1):235-42. DOI: 10.1111/j.1600-6143.2006.01585.x.
- Coelho T, Merlini G, Bulawa CE, et al. Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis. Neurol Ther 2016;5(1):1-25. DOI: 10.1007/s40120-016-0040-x.
- Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006;13(4):236-49. DOI: 10.1080/13506120600960882.
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013;310(24):2658-67. DOI: 10.1001/jama.2013.283815.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med 2018;379(11):1007-1016. DOI: 10.1056/NEJMoa1805689.
- Ackermann EJ, Guo S, Benson MD, et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid 2016;23(3):148-157. DOI: 10.1080/13506129.2016.1191458.
- Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 2013;369(9):819-29. DOI: 10.1056/NEJMoa1208760.
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379(1):22-31. DOI: 10.1056/NEJMoa1716793.
- Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med 2018;379(1):11-21. DOI: 10.1056/NEJMoa1716153.
- Adams D, Tournev IL, Taylor MS, et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 2023;30(1):1-9. DOI: 10.1080/13506129.2022.2091985.
- Coelho T, Marques W, Jr., Dasgupta NR, et al. Eplontersen for Hereditary Transthyretin Amyloidosis With Polyneuropathy. JAMA 2023;330(15):1448-1458. DOI: 10.1001/jama.2023.18688.
- Maurer MS, Kale P, Fontana M, et al. Patisiran Treatment in Patients with Transthyretin Cardiac Amyloidosis. N Engl J Med 2023;389(17):1553-1565. DOI: 10.1056/NEJMoa2300757.